 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMulti-state Protein Design with DynamicMPNN
Jul 29, 2025Structural biology has long been dominated by the one sequence, one structure, one function paradigm, yet many critical biological processes - from enzyme catalysis to membrane transport - depend on proteins that adopt multiple conformational states. Existing multi-state design approaches rely on post-hoc aggregation of single-state predictions, achieving poor experimental success rates compared to single-state design. We introduce DynamicMPNN, an inverse folding model explicitly trained to generate sequences compatible with multiple conformations through joint learning across conformational ensembles. Trained on 46,033 conformational pairs covering 75% of CATH superfamilies and evaluated using AlphaFold initial guess, DynamicMPNN outperforms ProteinMPNN by up to 13% on structure-normalized RMSD across our challenging multi-state protein benchmark.
All-atom Diffusion Transformers: Unified generative modelling of molecules and materials
Mar 05, 2025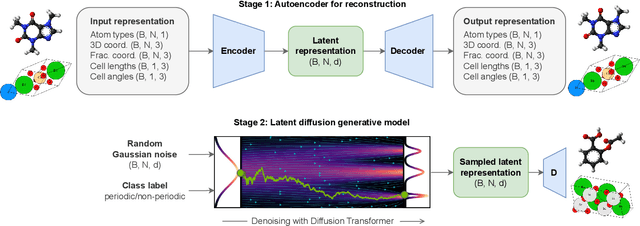

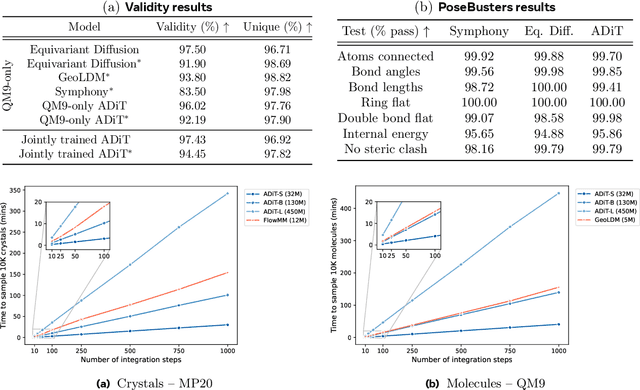
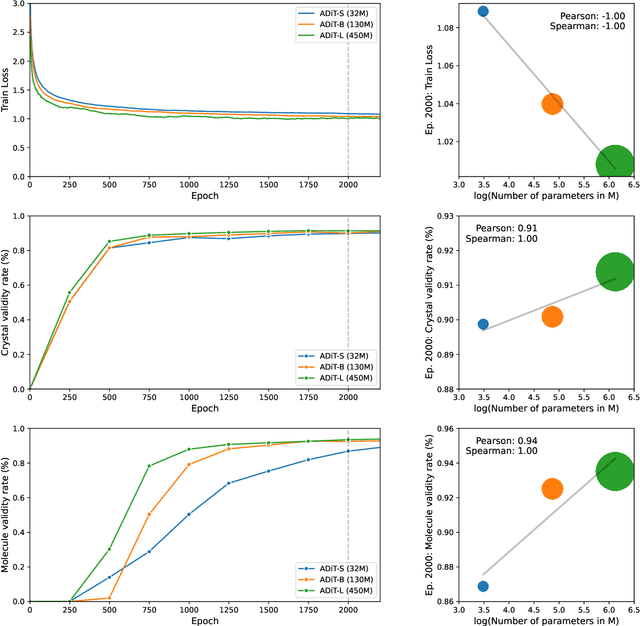
Diffusion models are the standard toolkit for generative modelling of 3D atomic systems. However, for different types of atomic systems - such as molecules and materials - the generative processes are usually highly specific to the target system despite the underlying physics being the same. We introduce the All-atom Diffusion Transformer (ADiT), a unified latent diffusion framework for jointly generating both periodic materials and non-periodic molecular systems using the same model: (1) An autoencoder maps a unified, all-atom representations of molecules and materials to a shared latent embedding space; and (2) A diffusion model is trained to generate new latent embeddings that the autoencoder can decode to sample new molecules or materials. Experiments on QM9 and MP20 datasets demonstrate that jointly trained ADiT generates realistic and valid molecules as well as materials, exceeding state-of-the-art results from molecule and crystal-specific models. ADiT uses standard Transformers for both the autoencoder and diffusion model, resulting in significant speedups during training and inference compared to equivariant diffusion models. Scaling ADiT up to half a billion parameters predictably improves performance, representing a step towards broadly generalizable foundation models for generative chemistry. Open source code: https://github.com/facebookresearch/all-atom-diffusion-transformer
Towards Mechanistic Interpretability of Graph Transformers via Attention Graphs
Feb 17, 2025We introduce Attention Graphs, a new tool for mechanistic interpretability of Graph Neural Networks (GNNs) and Graph Transformers based on the mathematical equivalence between message passing in GNNs and the self-attention mechanism in Transformers. Attention Graphs aggregate attention matrices across Transformer layers and heads to describe how information flows among input nodes. Through experiments on homophilous and heterophilous node classification tasks, we analyze Attention Graphs from a network science perspective and find that: (1) When Graph Transformers are allowed to learn the optimal graph structure using all-to-all attention among input nodes, the Attention Graphs learned by the model do not tend to correlate with the input/original graph structure; and (2) For heterophilous graphs, different Graph Transformer variants can achieve similar performance while utilising distinct information flow patterns. Open source code: https://github.com/batu-el/understanding-inductive-biases-of-gnns
Evaluating representation learning on the protein structure universe
Jun 19, 2024We introduce ProteinWorkshop, a comprehensive benchmark suite for representation learning on protein structures with Geometric Graph Neural Networks. We consider large-scale pre-training and downstream tasks on both experimental and predicted structures to enable the systematic evaluation of the quality of the learned structural representation and their usefulness in capturing functional relationships for downstream tasks. We find that: (1) large-scale pretraining on AlphaFold structures and auxiliary tasks consistently improve the performance of both rotation-invariant and equivariant GNNs, and (2) more expressive equivariant GNNs benefit from pretraining to a greater extent compared to invariant models. We aim to establish a common ground for the machine learning and computational biology communities to rigorously compare and advance protein structure representation learning. Our open-source codebase reduces the barrier to entry for working with large protein structure datasets by providing: (1) storage-efficient dataloaders for large-scale structural databases including AlphaFoldDB and ESM Atlas, as well as (2) utilities for constructing new tasks from the entire PDB. ProteinWorkshop is available at: github.com/a-r-j/ProteinWorkshop.
RNA-FrameFlow: Flow Matching for de novo 3D RNA Backbone Design
Jun 19, 2024We introduce RNA-FrameFlow, the first generative model for 3D RNA backbone design. We build upon SE(3) flow matching for protein backbone generation and establish protocols for data preparation and evaluation to address unique challenges posed by RNA modeling. We formulate RNA structures as a set of rigid-body frames and associated loss functions which account for larger, more conformationally flexible RNA backbones (13 atoms per nucleotide) vs. proteins (4 atoms per residue). Toward tackling the lack of diversity in 3D RNA datasets, we explore training with structural clustering and cropping augmentations. Additionally, we define a suite of evaluation metrics to measure whether the generated RNA structures are globally self-consistent (via inverse folding followed by forward folding) and locally recover RNA-specific structural descriptors. The most performant version of RNA-FrameFlow generates locally realistic RNA backbones of 40-150 nucleotides, over 40% of which pass our validity criteria as measured by a self-consistency TM-score >= 0.45, at which two RNAs have the same global fold. Open-source code: https://github.com/rish-16/rna-backbone-design
A Hitchhiker's Guide to Geometric GNNs for 3D Atomic Systems
Dec 12, 2023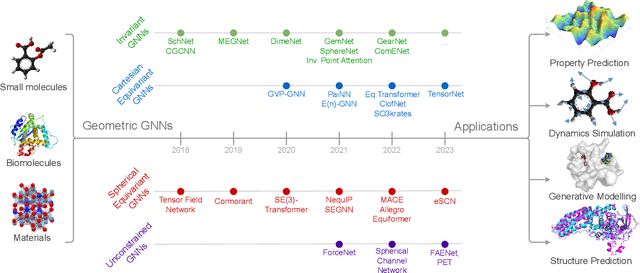
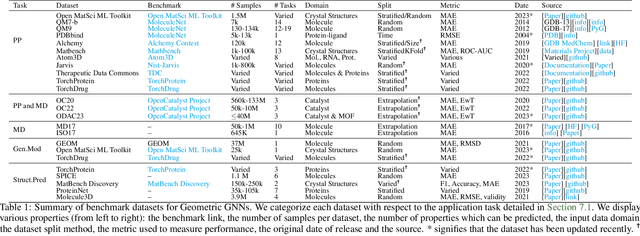
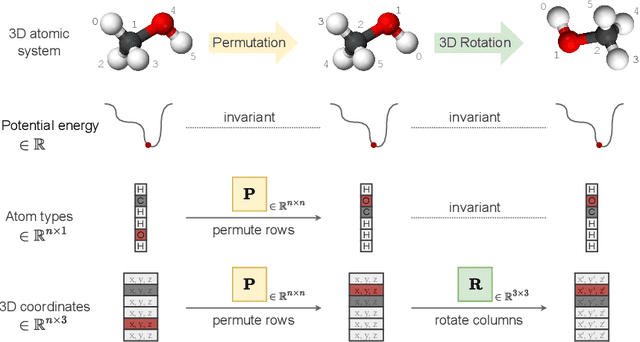
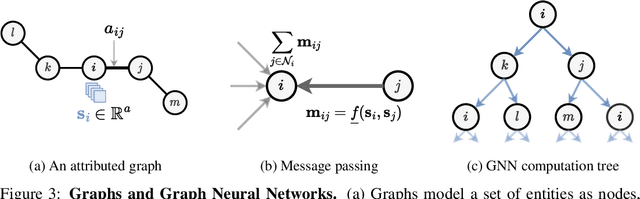
Recent advances in computational modelling of atomic systems, spanning molecules, proteins, and materials, represent them as geometric graphs with atoms embedded as nodes in 3D Euclidean space. In these graphs, the geometric attributes transform according to the inherent physical symmetries of 3D atomic systems, including rotations and translations in Euclidean space, as well as node permutations. In recent years, Geometric Graph Neural Networks have emerged as the preferred machine learning architecture powering applications ranging from protein structure prediction to molecular simulations and material generation. Their specificity lies in the inductive biases they leverage -- such as physical symmetries and chemical properties -- to learn informative representations of these geometric graphs. In this opinionated paper, we provide a comprehensive and self-contained overview of the field of Geometric GNNs for 3D atomic systems. We cover fundamental background material and introduce a pedagogical taxonomy of Geometric GNN architectures:(1) invariant networks, (2) equivariant networks in Cartesian basis, (3) equivariant networks in spherical basis, and (4) unconstrained networks. Additionally, we outline key datasets and application areas and suggest future research directions. The objective of this work is to present a structured perspective on the field, making it accessible to newcomers and aiding practitioners in gaining an intuition for its mathematical abstractions.
Artificial Intelligence for Science in Quantum, Atomistic, and Continuum Systems
Jul 17, 2023
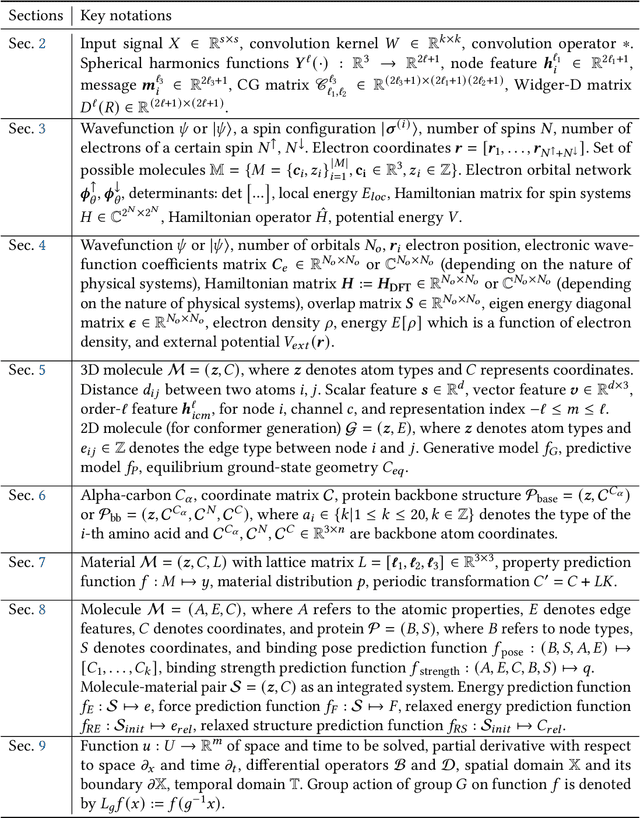
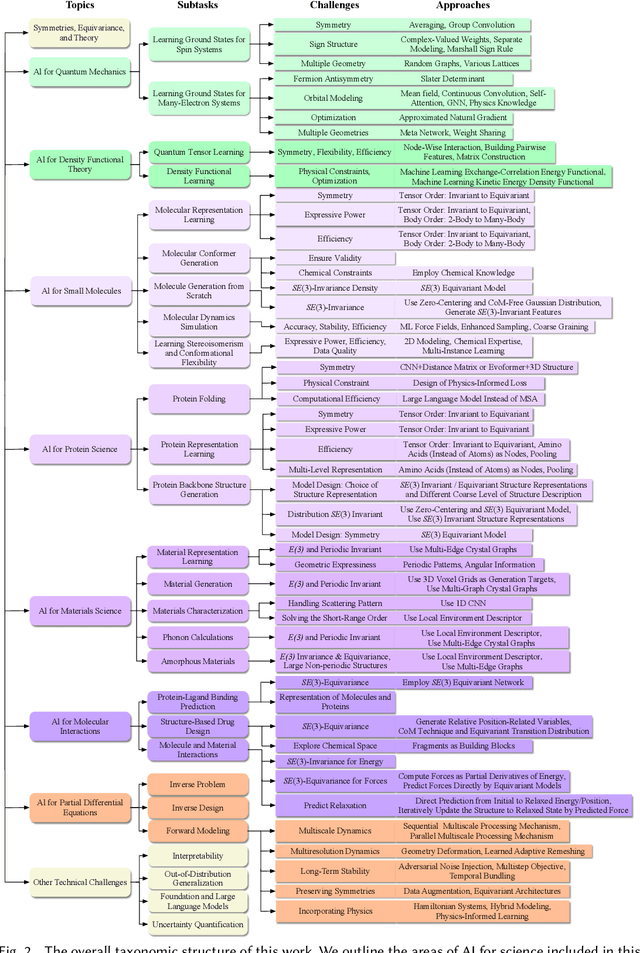

Advances in artificial intelligence (AI) are fueling a new paradigm of discoveries in natural sciences. Today, AI has started to advance natural sciences by improving, accelerating, and enabling our understanding of natural phenomena at a wide range of spatial and temporal scales, giving rise to a new area of research known as AI for science (AI4Science). Being an emerging research paradigm, AI4Science is unique in that it is an enormous and highly interdisciplinary area. Thus, a unified and technical treatment of this field is needed yet challenging. This paper aims to provide a technically thorough account of a subarea of AI4Science; namely, AI for quantum, atomistic, and continuum systems. These areas aim at understanding the physical world from the subatomic (wavefunctions and electron density), atomic (molecules, proteins, materials, and interactions), to macro (fluids, climate, and subsurface) scales and form an important subarea of AI4Science. A unique advantage of focusing on these areas is that they largely share a common set of challenges, thereby allowing a unified and foundational treatment. A key common challenge is how to capture physics first principles, especially symmetries, in natural systems by deep learning methods. We provide an in-depth yet intuitive account of techniques to achieve equivariance to symmetry transformations. We also discuss other common technical challenges, including explainability, out-of-distribution generalization, knowledge transfer with foundation and large language models, and uncertainty quantification. To facilitate learning and education, we provide categorized lists of resources that we found to be useful. We strive to be thorough and unified and hope this initial effort may trigger more community interests and efforts to further advance AI4Science.
Group Invariant Global Pooling
May 30, 2023

Much work has been devoted to devising architectures that build group-equivariant representations, while invariance is often induced using simple global pooling mechanisms. Little work has been done on creating expressive layers that are invariant to given symmetries, despite the success of permutation invariant pooling in various molecular tasks. In this work, we present Group Invariant Global Pooling (GIGP), an invariant pooling layer that is provably sufficiently expressive to represent a large class of invariant functions. We validate GIGP on rotated MNIST and QM9, showing improvements for the latter while attaining identical results for the former. By making the pooling process group orbit-aware, this invariant aggregation method leads to improved performance, while performing well-principled group aggregation.
Multi-State RNA Design with Geometric Multi-Graph Neural Networks
May 25, 2023Computational RNA design has broad applications across synthetic biology and therapeutic development. Fundamental to the diverse biological functions of RNA is its conformational flexibility, enabling single sequences to adopt a variety of distinct 3D states. Currently, computational biomolecule design tasks are often posed as inverse problems, where sequences are designed based on adopting a single desired structural conformation. In this work, we propose gRNAde, a geometric RNA design pipeline that operates on sets of 3D RNA backbone structures to explicitly account for and reflect RNA conformational diversity in its designs. We demonstrate the utility of gRNAde for improving native sequence recovery over single-state approaches on a new large-scale 3D RNA design dataset, especially for multi-state and structurally diverse RNAs. Our code is available at https://github.com/chaitjo/geometric-rna-design
On the Expressive Power of Geometric Graph Neural Networks
Jan 23, 2023The expressive power of Graph Neural Networks (GNNs) has been studied extensively through the Weisfeiler-Leman (WL) graph isomorphism test. However, standard GNNs and the WL framework are inapplicable for geometric graphs embedded in Euclidean space, such as biomolecules, materials, and other physical systems. In this work, we propose a geometric version of the WL test (GWL) for discriminating geometric graphs while respecting the underlying physical symmetries: permutations, rotation, reflection, and translation. We use GWL to characterise the expressive power of geometric GNNs that are invariant or equivariant to physical symmetries in terms of distinguishing geometric graphs. GWL unpacks how key design choices influence geometric GNN expressivity: (1) Invariant layers have limited expressivity as they cannot distinguish one-hop identical geometric graphs; (2) Equivariant layers distinguish a larger class of graphs by propagating geometric information beyond local neighbourhoods; (3) Higher order tensors and scalarisation enable maximally powerful geometric GNNs; and (4) GWL's discrimination-based perspective is equivalent to universal approximation. Synthetic experiments supplementing our results are available at https://github.com/chaitjo/geometric-gnn-dojo