 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTriForces: Augmenting Atomistic GNNs for Transferable Representations
May 20, 2026Machine learning interatomic potentials (MLIPs) achieve excellent accuracy when trained on large Density Functional Theory (DFT) data. To be useful in practice, they must often be adapted to target chemistries using small and expensive task-specific datasets. However, MLIPs transfer inconsistently across domains, with representations that often loose accessible composition and structure information. To address this, we present TriForces, a model-agnostic three-stream framework that separates composition and structure information, combined with self-supervised learning to preserve transferable representations. TriForces improves performance on MatBench and QM9 over baselines without needing DFT labels and enables efficient similar structure retrieval through its learned latent space. On OMat24, in limited-data training regime, TriForces reduces energy MAE by 57% at 20K samples only and improves force MAE across sample sizes. We release pretrained TriForces variants across multiple MLIP architectures with code at https://github.com/Ramlaoui/triforces.
Catalyst GFlowNet for electrocatalyst design: A hydrogen evolution reaction case study
Oct 02, 2025Efficient and inexpensive energy storage is essential for accelerating the adoption of renewable energy and ensuring a stable supply, despite fluctuations in sources such as wind and solar. Electrocatalysts play a key role in hydrogen energy storage (HES), allowing the energy to be stored as hydrogen. However, the development of affordable and high-performance catalysts for this process remains a significant challenge. We introduce Catalyst GFlowNet, a generative model that leverages machine learning-based predictors of formation and adsorption energy to design crystal surfaces that act as efficient catalysts. We demonstrate the performance of the model through a proof-of-concept application to the hydrogen evolution reaction, a key reaction in HES, for which we successfully identified platinum as the most efficient known catalyst. In future work, we aim to extend this approach to the oxygen evolution reaction, where current optimal catalysts are expensive metal oxides, and open the search space to discover new materials. This generative modeling framework offers a promising pathway for accelerating the search for novel and efficient catalysts.
LeMat-Traj: A Scalable and Unified Dataset of Materials Trajectories for Atomistic Modeling
Aug 28, 2025



The development of accurate machine learning interatomic potentials (MLIPs) is limited by the fragmented availability and inconsistent formatting of quantum mechanical trajectory datasets derived from Density Functional Theory (DFT). These datasets are expensive to generate yet difficult to combine due to variations in format, metadata, and accessibility. To address this, we introduce LeMat-Traj, a curated dataset comprising over 120 million atomic configurations aggregated from large-scale repositories, including the Materials Project, Alexandria, and OQMD. LeMat-Traj standardizes data representation, harmonizes results and filters for high-quality configurations across widely used DFT functionals (PBE, PBESol, SCAN, r2SCAN). It significantly lowers the barrier for training transferrable and accurate MLIPs. LeMat-Traj spans both relaxed low-energy states and high-energy, high-force structures, complementing molecular dynamics and active learning datasets. By fine-tuning models pre-trained on high-force data with LeMat-Traj, we achieve a significant reduction in force prediction errors on relaxation tasks. We also present LeMaterial-Fetcher, a modular and extensible open-source library developed for this work, designed to provide a reproducible framework for the community to easily incorporate new data sources and ensure the continued evolution of large-scale materials datasets. LeMat-Traj and LeMaterial-Fetcher are publicly available at https://huggingface.co/datasets/LeMaterial/LeMat-Traj and https://github.com/LeMaterial/lematerial-fetcher.
Improving Molecular Modeling with Geometric GNNs: an Empirical Study
Jul 11, 2024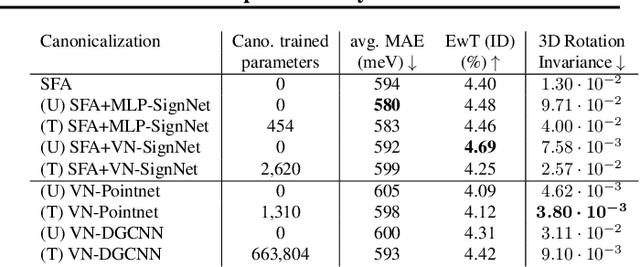


Rapid advancements in machine learning (ML) are transforming materials science by significantly speeding up material property calculations. However, the proliferation of ML approaches has made it challenging for scientists to keep up with the most promising techniques. This paper presents an empirical study on Geometric Graph Neural Networks for 3D atomic systems, focusing on the impact of different (1) canonicalization methods, (2) graph creation strategies, and (3) auxiliary tasks, on performance, scalability and symmetry enforcement. Our findings and insights aim to guide researchers in selecting optimal modeling components for molecular modeling tasks.
A Hitchhiker's Guide to Geometric GNNs for 3D Atomic Systems
Dec 12, 2023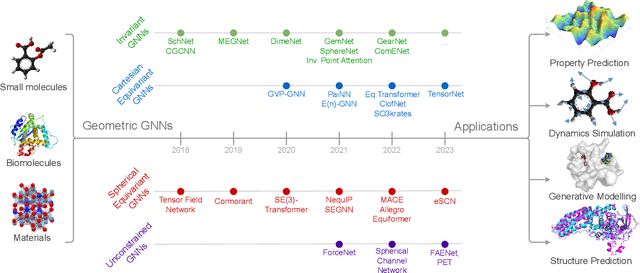
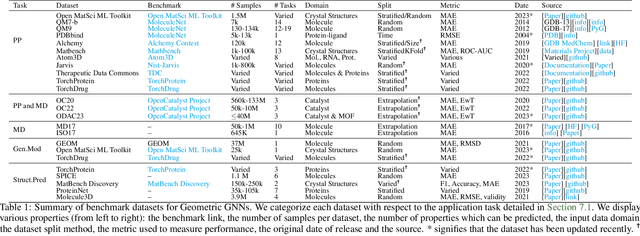
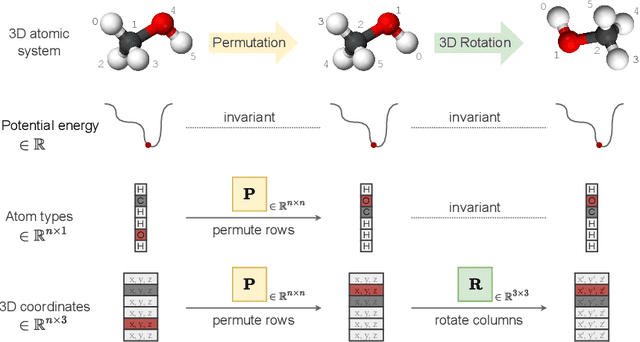
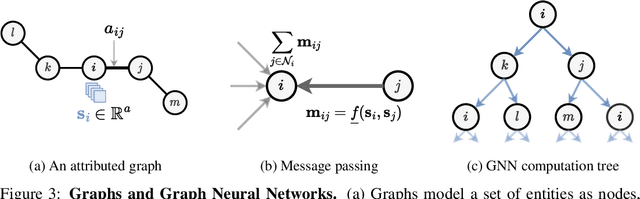
Recent advances in computational modelling of atomic systems, spanning molecules, proteins, and materials, represent them as geometric graphs with atoms embedded as nodes in 3D Euclidean space. In these graphs, the geometric attributes transform according to the inherent physical symmetries of 3D atomic systems, including rotations and translations in Euclidean space, as well as node permutations. In recent years, Geometric Graph Neural Networks have emerged as the preferred machine learning architecture powering applications ranging from protein structure prediction to molecular simulations and material generation. Their specificity lies in the inductive biases they leverage -- such as physical symmetries and chemical properties -- to learn informative representations of these geometric graphs. In this opinionated paper, we provide a comprehensive and self-contained overview of the field of Geometric GNNs for 3D atomic systems. We cover fundamental background material and introduce a pedagogical taxonomy of Geometric GNN architectures:(1) invariant networks, (2) equivariant networks in Cartesian basis, (3) equivariant networks in spherical basis, and (4) unconstrained networks. Additionally, we outline key datasets and application areas and suggest future research directions. The objective of this work is to present a structured perspective on the field, making it accessible to newcomers and aiding practitioners in gaining an intuition for its mathematical abstractions.
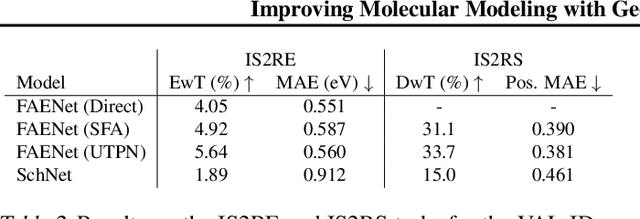
On the importance of catalyst-adsorbate 3D interactions for relaxed energy predictions
Oct 10, 2023The use of machine learning for material property prediction and discovery has traditionally centered on graph neural networks that incorporate the geometric configuration of all atoms. However, in practice not all this information may be readily available, e.g.~when evaluating the potentially unknown binding of adsorbates to catalyst. In this paper, we investigate whether it is possible to predict a system's relaxed energy in the OC20 dataset while ignoring the relative position of the adsorbate with respect to the electro-catalyst. We consider SchNet, DimeNet++ and FAENet as base architectures and measure the impact of four modifications on model performance: removing edges in the input graph, pooling independent representations, not sharing the backbone weights and using an attention mechanism to propagate non-geometric relative information. We find that while removing binding site information impairs accuracy as expected, modified models are able to predict relaxed energies with remarkably decent MAE. Our work suggests future research directions in accelerated materials discovery where information on reactant configurations can be reduced or altogether omitted.
Crystal-GFN: sampling crystals with desirable properties and constraints
Oct 07, 2023Accelerating material discovery holds the potential to greatly help mitigate the climate crisis. Discovering new solid-state crystals such as electrocatalysts, ionic conductors or photovoltaics can have a crucial impact, for instance, in improving the efficiency of renewable energy production and storage. In this paper, we introduce Crystal-GFlowNet, a generative model of crystal structures that sequentially samples a crystal's composition, space group and lattice parameters. This domain-inspired approach enables the flexible incorporation of physical and geometrical constraints, as well as the use of any available predictive model of a desired property as an objective function. We evaluate the capabilities of Crystal-GFlowNet by using as objective the formation energy of a crystal structure, as predicted by a new proxy model trained on MatBench. The results demonstrate that Crystal-GFlowNet is able to sample diverse crystals with low formation energy.
torchgfn: A PyTorch GFlowNet library
May 24, 2023
The increasing popularity of generative flow networks (GFlowNets or GFNs) is accompanied with a proliferation of code sources. This hinders the implementation of new features, such as training losses, that can readily be compared to existing ones, on a set of common environments. In addition to slowing down research in the field of GFlowNets, different code bases use different conventions, that might be confusing for newcomers. `torchgfn` is a library built on top of PyTorch, that aims at addressing both problems. It provides user with a simple API for environments, and useful abstractions for samplers and losses. Multiple examples are provided, replicating published results. The code is available in https://github.com/saleml/torchgfn.
FAENet: Frame Averaging Equivariant GNN for Materials Modeling
Apr 28, 2023Applications of machine learning techniques for materials modeling typically involve functions known to be equivariant or invariant to specific symmetries. While graph neural networks (GNNs) have proven successful in such tasks, they enforce symmetries via the model architecture, which often reduces their expressivity, scalability and comprehensibility. In this paper, we introduce (1) a flexible framework relying on stochastic frame-averaging (SFA) to make any model E(3)-equivariant or invariant through data transformations. (2) FAENet: a simple, fast and expressive GNN, optimized for SFA, that processes geometric information without any symmetrypreserving design constraints. We prove the validity of our method theoretically and empirically demonstrate its superior accuracy and computational scalability in materials modeling on the OC20 dataset (S2EF, IS2RE) as well as common molecular modeling tasks (QM9, QM7-X). A package implementation is available at https://faenet.readthedocs.io.
PhAST: Physics-Aware, Scalable, and Task-specific GNNs for Accelerated Catalyst Design
Nov 22, 2022Mitigating the climate crisis requires a rapid transition towards lower carbon energy. Catalyst materials play a crucial role in the electrochemical reactions involved in a great number of industrial processes key to this transition, such as renewable energy storage and electrofuel synthesis. To reduce the amount of energy spent on such processes, we must quickly discover more efficient catalysts to drive the electrochemical reactions. Machine learning (ML) holds the potential to efficiently model the properties of materials from large amounts of data, and thus to accelerate electrocatalyst design. The Open Catalyst Project OC20 data set was constructed to that end. However, most existing ML models trained on OC20 are still neither scalable nor accurate enough for practical applications. Here, we propose several task-specific innovations, applicable to most architectures, which increase both computational efficiency and accuracy. In particular, we propose improvements in (1) the graph creation step, (2) atom representations and (3) the energy prediction head. We describe these contributions and evaluate them on several architectures, showing up to 5$\times$ reduction in inference time without sacrificing accuracy.