Get our free extension to see links to code for papers anywhere online!Free add-on: code for papers everywhere!Free add-on: See code for papers anywhere!
 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImproving Molecular Modeling with Geometric GNNs: an Empirical Study
Paper and Code
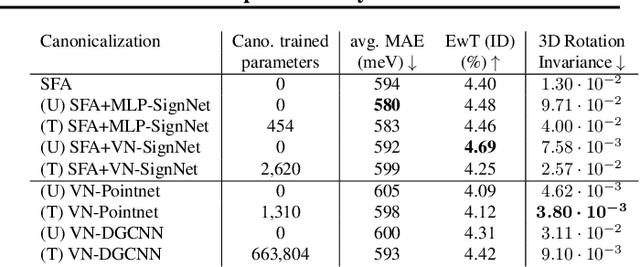

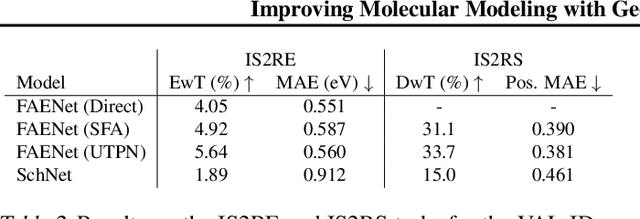

Rapid advancements in machine learning (ML) are transforming materials science by significantly speeding up material property calculations. However, the proliferation of ML approaches has made it challenging for scientists to keep up with the most promising techniques. This paper presents an empirical study on Geometric Graph Neural Networks for 3D atomic systems, focusing on the impact of different (1) canonicalization methods, (2) graph creation strategies, and (3) auxiliary tasks, on performance, scalability and symmetry enforcement. Our findings and insights aim to guide researchers in selecting optimal modeling components for molecular modeling tasks.
View paper on