 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCatalyst GFlowNet for electrocatalyst design: A hydrogen evolution reaction case study
Oct 02, 2025Efficient and inexpensive energy storage is essential for accelerating the adoption of renewable energy and ensuring a stable supply, despite fluctuations in sources such as wind and solar. Electrocatalysts play a key role in hydrogen energy storage (HES), allowing the energy to be stored as hydrogen. However, the development of affordable and high-performance catalysts for this process remains a significant challenge. We introduce Catalyst GFlowNet, a generative model that leverages machine learning-based predictors of formation and adsorption energy to design crystal surfaces that act as efficient catalysts. We demonstrate the performance of the model through a proof-of-concept application to the hydrogen evolution reaction, a key reaction in HES, for which we successfully identified platinum as the most efficient known catalyst. In future work, we aim to extend this approach to the oxygen evolution reaction, where current optimal catalysts are expensive metal oxides, and open the search space to discover new materials. This generative modeling framework offers a promising pathway for accelerating the search for novel and efficient catalysts.
LeMat-Traj: A Scalable and Unified Dataset of Materials Trajectories for Atomistic Modeling
Aug 28, 2025The development of accurate machine learning interatomic potentials (MLIPs) is limited by the fragmented availability and inconsistent formatting of quantum mechanical trajectory datasets derived from Density Functional Theory (DFT). These datasets are expensive to generate yet difficult to combine due to variations in format, metadata, and accessibility. To address this, we introduce LeMat-Traj, a curated dataset comprising over 120 million atomic configurations aggregated from large-scale repositories, including the Materials Project, Alexandria, and OQMD. LeMat-Traj standardizes data representation, harmonizes results and filters for high-quality configurations across widely used DFT functionals (PBE, PBESol, SCAN, r2SCAN). It significantly lowers the barrier for training transferrable and accurate MLIPs. LeMat-Traj spans both relaxed low-energy states and high-energy, high-force structures, complementing molecular dynamics and active learning datasets. By fine-tuning models pre-trained on high-force data with LeMat-Traj, we achieve a significant reduction in force prediction errors on relaxation tasks. We also present LeMaterial-Fetcher, a modular and extensible open-source library developed for this work, designed to provide a reproducible framework for the community to easily incorporate new data sources and ensure the continued evolution of large-scale materials datasets. LeMat-Traj and LeMaterial-Fetcher are publicly available at https://huggingface.co/datasets/LeMaterial/LeMat-Traj and https://github.com/LeMaterial/lematerial-fetcher.
Improving Molecular Modeling with Geometric GNNs: an Empirical Study
Jul 11, 2024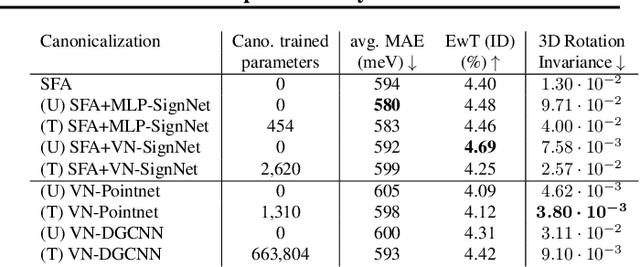

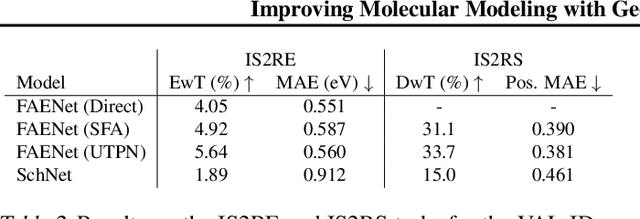

Rapid advancements in machine learning (ML) are transforming materials science by significantly speeding up material property calculations. However, the proliferation of ML approaches has made it challenging for scientists to keep up with the most promising techniques. This paper presents an empirical study on Geometric Graph Neural Networks for 3D atomic systems, focusing on the impact of different (1) canonicalization methods, (2) graph creation strategies, and (3) auxiliary tasks, on performance, scalability and symmetry enforcement. Our findings and insights aim to guide researchers in selecting optimal modeling components for molecular modeling tasks.