 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTriForces: Augmenting Atomistic GNNs for Transferable Representations
May 20, 2026Machine learning interatomic potentials (MLIPs) achieve excellent accuracy when trained on large Density Functional Theory (DFT) data. To be useful in practice, they must often be adapted to target chemistries using small and expensive task-specific datasets. However, MLIPs transfer inconsistently across domains, with representations that often loose accessible composition and structure information. To address this, we present TriForces, a model-agnostic three-stream framework that separates composition and structure information, combined with self-supervised learning to preserve transferable representations. TriForces improves performance on MatBench and QM9 over baselines without needing DFT labels and enables efficient similar structure retrieval through its learned latent space. On OMat24, in limited-data training regime, TriForces reduces energy MAE by 57% at 20K samples only and improves force MAE across sample sizes. We release pretrained TriForces variants across multiple MLIP architectures with code at https://github.com/Ramlaoui/triforces.
LeMat-Traj: A Scalable and Unified Dataset of Materials Trajectories for Atomistic Modeling
Aug 28, 2025



The development of accurate machine learning interatomic potentials (MLIPs) is limited by the fragmented availability and inconsistent formatting of quantum mechanical trajectory datasets derived from Density Functional Theory (DFT). These datasets are expensive to generate yet difficult to combine due to variations in format, metadata, and accessibility. To address this, we introduce LeMat-Traj, a curated dataset comprising over 120 million atomic configurations aggregated from large-scale repositories, including the Materials Project, Alexandria, and OQMD. LeMat-Traj standardizes data representation, harmonizes results and filters for high-quality configurations across widely used DFT functionals (PBE, PBESol, SCAN, r2SCAN). It significantly lowers the barrier for training transferrable and accurate MLIPs. LeMat-Traj spans both relaxed low-energy states and high-energy, high-force structures, complementing molecular dynamics and active learning datasets. By fine-tuning models pre-trained on high-force data with LeMat-Traj, we achieve a significant reduction in force prediction errors on relaxation tasks. We also present LeMaterial-Fetcher, a modular and extensible open-source library developed for this work, designed to provide a reproducible framework for the community to easily incorporate new data sources and ensure the continued evolution of large-scale materials datasets. LeMat-Traj and LeMaterial-Fetcher are publicly available at https://huggingface.co/datasets/LeMaterial/LeMat-Traj and https://github.com/LeMaterial/lematerial-fetcher.
Rotationally Invariant Latent Distances for Uncertainty Estimation of Relaxed Energy Predictions by Graph Neural Network Potentials
Jul 15, 2024Graph neural networks (GNNs) have been shown to be astonishingly capable models for molecular property prediction, particularly as surrogates for expensive density functional theory calculations of relaxed energy for novel material discovery. However, one limitation of GNNs in this context is the lack of useful uncertainty prediction methods, as this is critical to the material discovery pipeline. In this work, we show that uncertainty quantification for relaxed energy calculations is more complex than uncertainty quantification for other kinds of molecular property prediction, due to the effect that structure optimizations have on the error distribution. We propose that distribution-free techniques are more useful tools for assessing calibration, recalibrating, and developing uncertainty prediction methods for GNNs performing relaxed energy calculations. We also develop a relaxed energy task for evaluating uncertainty methods for equivariant GNNs, based on distribution-free recalibration and using the Open Catalyst Project dataset. We benchmark a set of popular uncertainty prediction methods on this task, and show that latent distance methods, with our novel improvements, are the most well-calibrated and economical approach for relaxed energy calculations. Finally, we demonstrate that our latent space distance method produces results which align with our expectations on a clustering example, and on specific equation of state and adsorbate coverage examples from outside the training dataset.
FINETUNA: Fine-tuning Accelerated Molecular Simulations
May 02, 2022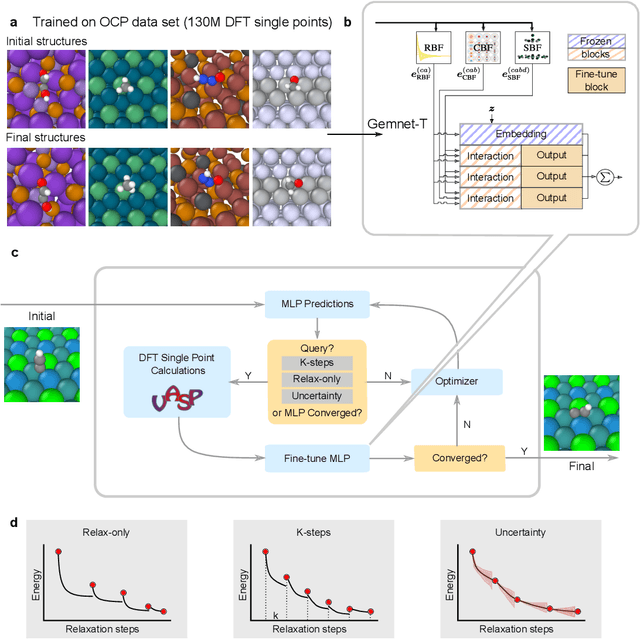
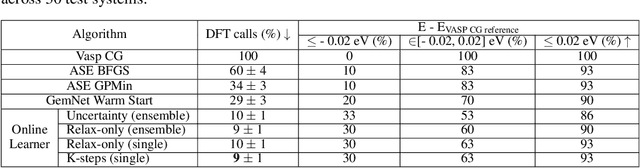
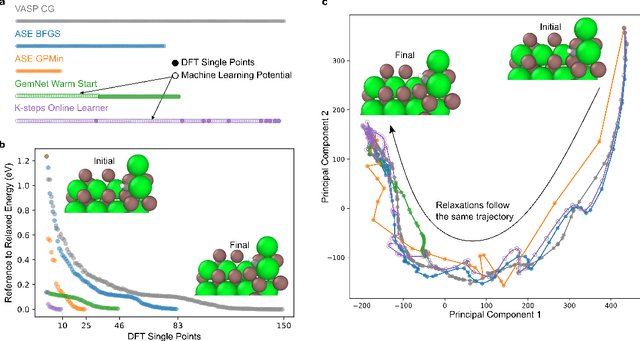
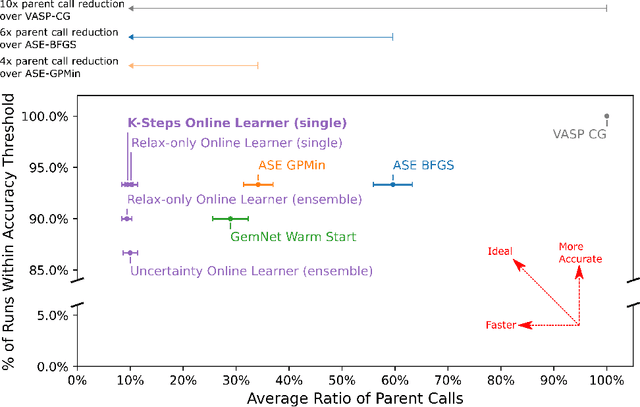
Machine learning approaches have the potential to approximate Density Functional Theory (DFT) for atomistic simulations in a computationally efficient manner, which could dramatically increase the impact of computational simulations on real-world problems. However, they are limited by their accuracy and the cost of generating labeled data. Here, we present an online active learning framework for accelerating the simulation of atomic systems efficiently and accurately by incorporating prior physical information learned by large-scale pre-trained graph neural network models from the Open Catalyst Project. Accelerating these simulations enables useful data to be generated more cheaply, allowing better models to be trained and more atomistic systems to be screened. We also present a method of comparing local optimization techniques on the basis of both their speed and accuracy. Experiments on 30 benchmark adsorbate-catalyst systems show that our method of transfer learning to incorporate prior information from pre-trained models accelerates simulations by reducing the number of DFT calculations by 91%, while meeting an accuracy threshold of 0.02 eV 93% of the time. Finally, we demonstrate a technique for leveraging the interactive functionality built in to VASP to efficiently compute single point calculations within our online active learning framework without the significant startup costs. This allows VASP to work in tandem with our framework while requiring 75% fewer self-consistent cycles than conventional single point calculations. The online active learning implementation, and examples using the VASP interactive code, are available in the open source FINETUNA package on Github.