 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFine-Tuned Language Models Generate Stable Inorganic Materials as Text
Feb 06, 2024We propose fine-tuning large language models for generation of stable materials. While unorthodox, fine-tuning large language models on text-encoded atomistic data is simple to implement yet reliable, with around 90% of sampled structures obeying physical constraints on atom positions and charges. Using energy above hull calculations from both learned ML potentials and gold-standard DFT calculations, we show that our strongest model (fine-tuned LLaMA-2 70B) can generate materials predicted to be metastable at about twice the rate (49% vs 28%) of CDVAE, a competing diffusion model. Because of text prompting's inherent flexibility, our models can simultaneously be used for unconditional generation of stable material, infilling of partial structures and text-conditional generation. Finally, we show that language models' ability to capture key symmetries of crystal structures improves with model scale, suggesting that the biases of pretrained LLMs are surprisingly well-suited for atomistic data.
The Open DAC 2023 Dataset and Challenges for Sorbent Discovery in Direct Air Capture
Nov 01, 2023
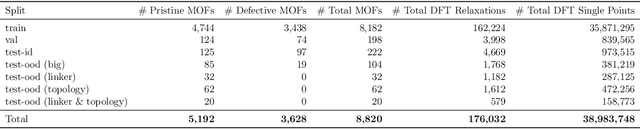
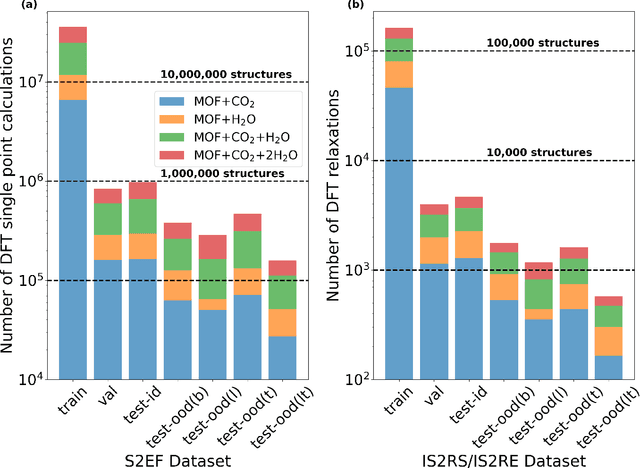

New methods for carbon dioxide removal are urgently needed to combat global climate change. Direct air capture (DAC) is an emerging technology to capture carbon dioxide directly from ambient air. Metal-organic frameworks (MOFs) have been widely studied as potentially customizable adsorbents for DAC. However, discovering promising MOF sorbents for DAC is challenging because of the vast chemical space to explore and the need to understand materials as functions of humidity and temperature. We explore a computational approach benefiting from recent innovations in machine learning (ML) and present a dataset named Open DAC 2023 (ODAC23) consisting of more than 38M density functional theory (DFT) calculations on more than 8,800 MOF materials containing adsorbed CO2 and/or H2O. ODAC23 is by far the largest dataset of MOF adsorption calculations at the DFT level of accuracy currently available. In addition to probing properties of adsorbed molecules, the dataset is a rich source of information on structural relaxation of MOFs, which will be useful in many contexts beyond specific applications for DAC. A large number of MOFs with promising properties for DAC are identified directly in ODAC23. We also trained state-of-the-art ML models on this dataset to approximate calculations at the DFT level. This open-source dataset and our initial ML models will provide an important baseline for future efforts to identify MOFs for a wide range of applications, including DAC.
Spherical Channels for Modeling Atomic Interactions
Jun 29, 2022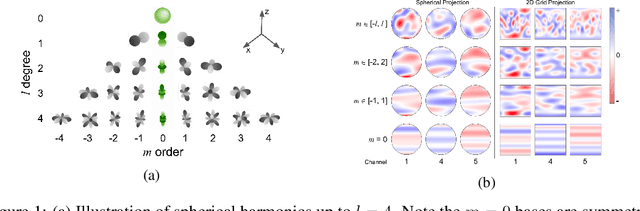
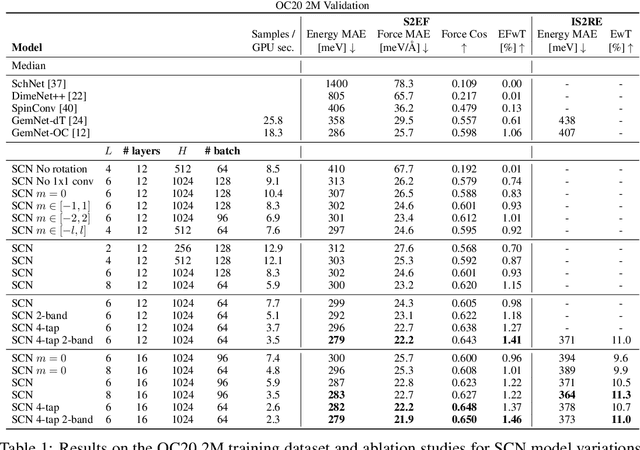
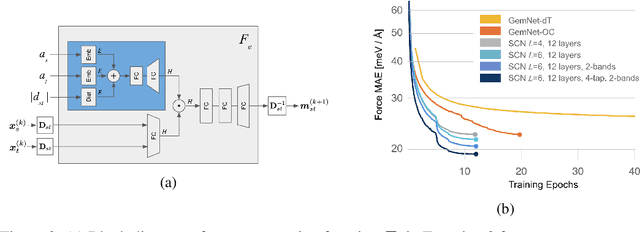
Modeling the energy and forces of atomic systems is a fundamental problem in computational chemistry with the potential to help address many of the world's most pressing problems, including those related to energy scarcity and climate change. These calculations are traditionally performed using Density Functional Theory, which is computationally very expensive. Machine learning has the potential to dramatically improve the efficiency of these calculations from days or hours to seconds. We propose the Spherical Channel Network (SCN) to model atomic energies and forces. The SCN is a graph neural network where nodes represent atoms and edges their neighboring atoms. The atom embeddings are a set of spherical functions, called spherical channels, represented using spherical harmonics. We demonstrate, that by rotating the embeddings based on the 3D edge orientation, more information may be utilized while maintaining the rotational equivariance of the messages. While equivariance is a desirable property, we find that by relaxing this constraint in both message passing and aggregation, improved accuracy may be achieved. We demonstrate state-of-the-art results on the large-scale Open Catalyst 2020 dataset in both energy and force prediction for numerous tasks and metrics.
The Open Catalyst 2022 Dataset and Challenges for Oxide Electrocatalysis
Jun 17, 2022
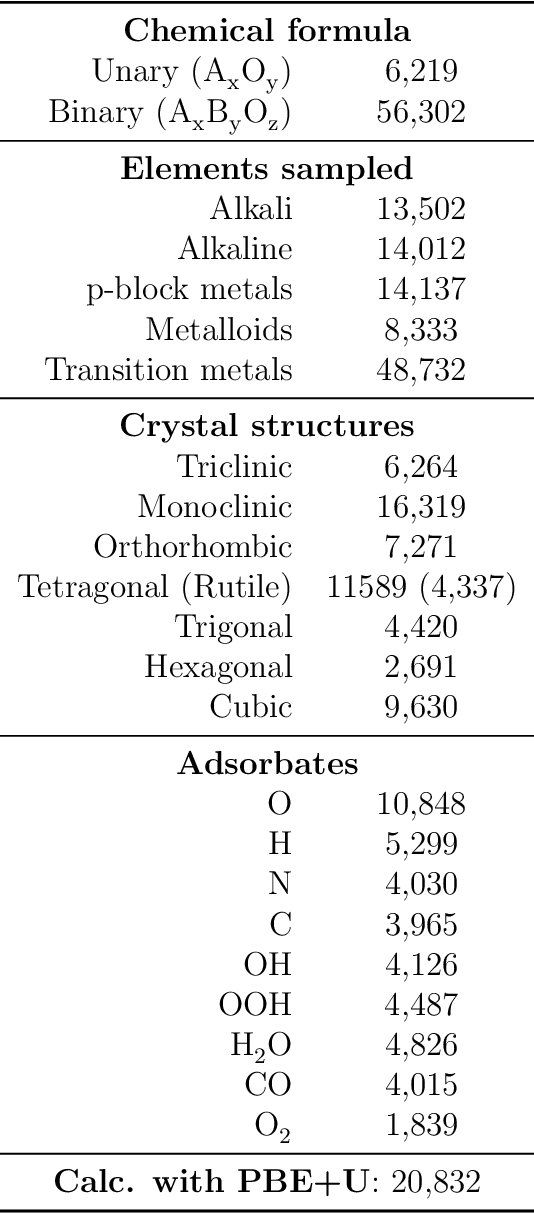
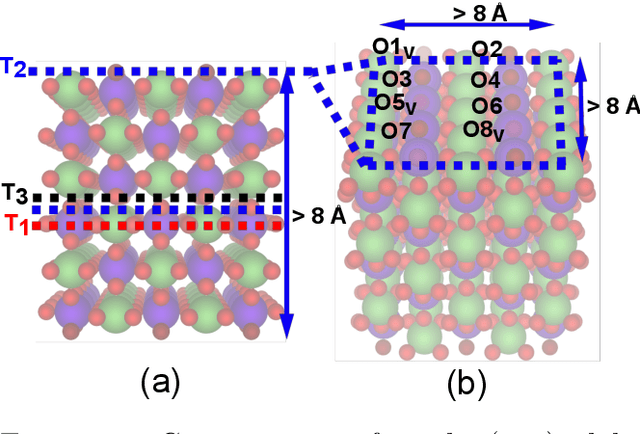
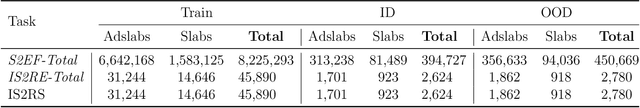
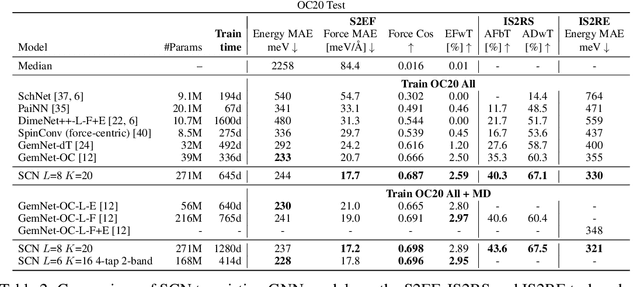
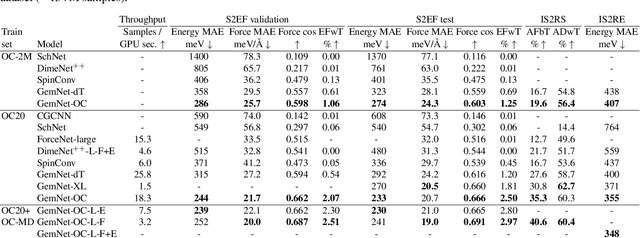
Computational catalysis and machine learning communities have made considerable progress in developing machine learning models for catalyst discovery and design. Yet, a general machine learning potential that spans the chemical space of catalysis is still out of reach. A significant hurdle is obtaining access to training data across a wide range of materials. One important class of materials where data is lacking are oxides, which inhibits models from studying the Oxygen Evolution Reaction and oxide electrocatalysis more generally. To address this we developed the Open Catalyst 2022(OC22) dataset, consisting of 62,521 Density Functional Theory (DFT) relaxations (~9,884,504 single point calculations) across a range of oxide materials, coverages, and adsorbates (*H, *O, *N, *C, *OOH, *OH, *OH2, *O2, *CO). We define generalized tasks to predict the total system energy that are applicable across catalysis, develop baseline performance of several graph neural networks (SchNet, DimeNet++, ForceNet, SpinConv, PaiNN, GemNet-dT, GemNet-OC), and provide pre-defined dataset splits to establish clear benchmarks for future efforts. For all tasks, we study whether combining datasets leads to better results, even if they contain different materials or adsorbates. Specifically, we jointly train models on Open Catalyst 2020 (OC20) Dataset and OC22, or fine-tune pretrained OC20 models on OC22. In the most general task, GemNet-OC sees a ~32% improvement in energy predictions through fine-tuning and a ~9% improvement in force predictions via joint training. Surprisingly, joint training on both the OC20 and much smaller OC22 datasets also improves total energy predictions on OC20 by ~19%. The dataset and baseline models are open sourced, and a public leaderboard will follow to encourage continued community developments on the total energy tasks and data.
FINETUNA: Fine-tuning Accelerated Molecular Simulations
May 02, 2022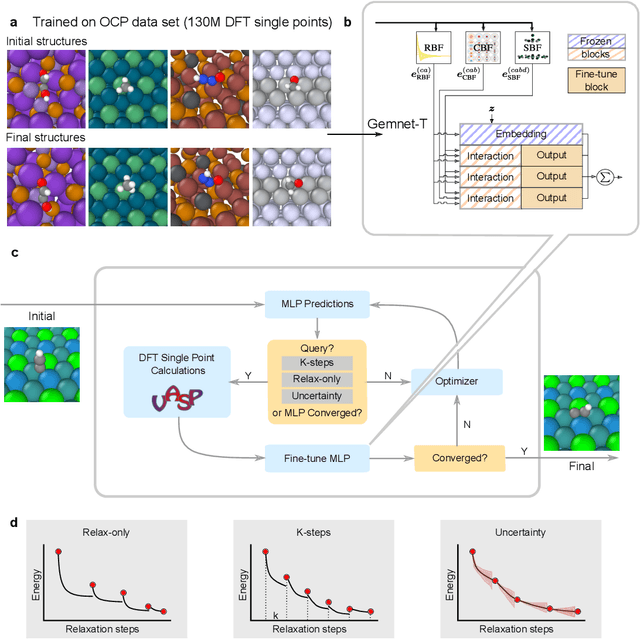
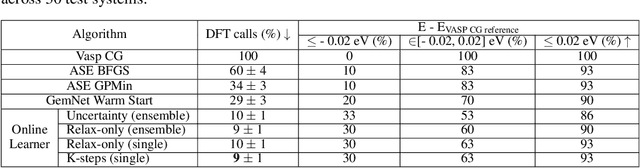
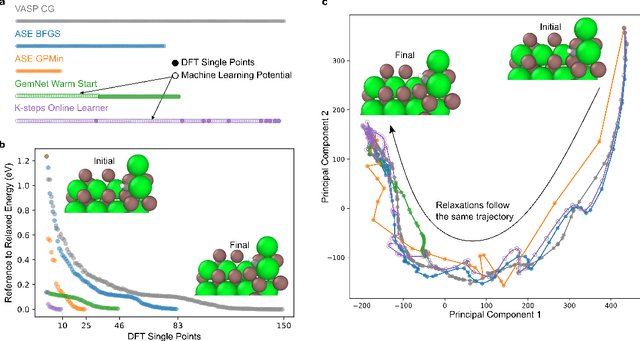
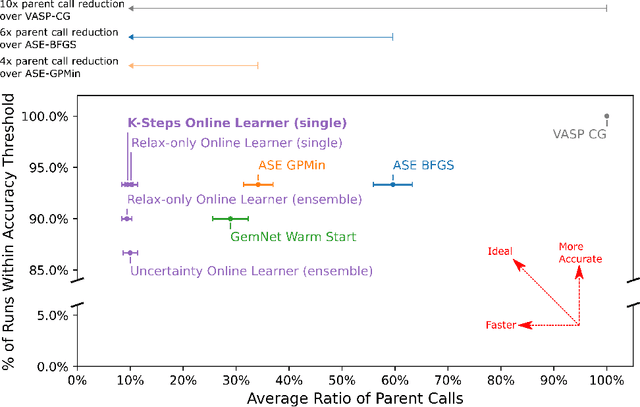
Machine learning approaches have the potential to approximate Density Functional Theory (DFT) for atomistic simulations in a computationally efficient manner, which could dramatically increase the impact of computational simulations on real-world problems. However, they are limited by their accuracy and the cost of generating labeled data. Here, we present an online active learning framework for accelerating the simulation of atomic systems efficiently and accurately by incorporating prior physical information learned by large-scale pre-trained graph neural network models from the Open Catalyst Project. Accelerating these simulations enables useful data to be generated more cheaply, allowing better models to be trained and more atomistic systems to be screened. We also present a method of comparing local optimization techniques on the basis of both their speed and accuracy. Experiments on 30 benchmark adsorbate-catalyst systems show that our method of transfer learning to incorporate prior information from pre-trained models accelerates simulations by reducing the number of DFT calculations by 91%, while meeting an accuracy threshold of 0.02 eV 93% of the time. Finally, we demonstrate a technique for leveraging the interactive functionality built in to VASP to efficiently compute single point calculations within our online active learning framework without the significant startup costs. This allows VASP to work in tandem with our framework while requiring 75% fewer self-consistent cycles than conventional single point calculations. The online active learning implementation, and examples using the VASP interactive code, are available in the open source FINETUNA package on Github.
How Do Graph Networks Generalize to Large and Diverse Molecular Systems?
Apr 06, 2022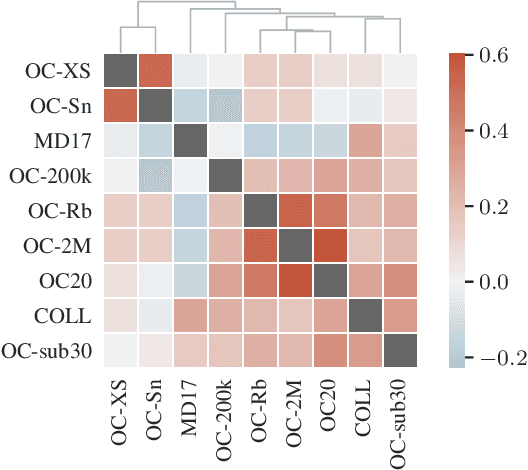
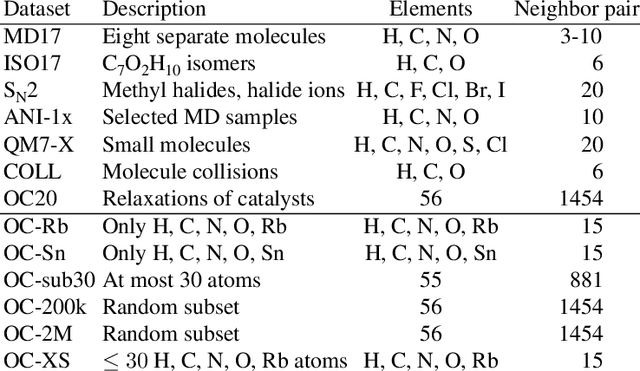
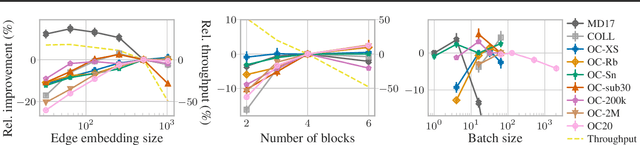
The predominant method of demonstrating progress of atomic graph neural networks are benchmarks on small and limited datasets. The implicit hypothesis behind this approach is that progress on these narrow datasets generalize to the large diversity of chemistry. This generalizability would be very helpful for research, but currently remains untested. In this work we test this assumption by identifying four aspects of complexity in which many datasets are lacking: 1. Chemical diversity (number of different elements), 2. system size (number of atoms per sample), 3. dataset size (number of data samples), and 4. domain shift (similarity of the training and test set). We introduce multiple subsets of the large Open Catalyst 2020 (OC20) dataset to independently investigate each of these aspects. We then perform 21 ablation studies and sensitivity analyses on 9 datasets testing both previously proposed and new model enhancements. We find that some improvements are consistent between datasets, but many are not and some even have opposite effects. Based on this analysis, we identify a smaller dataset that correlates well with the full OC20 dataset, and propose the GemNet-OC model, which outperforms the previous state-of-the-art on OC20 by 16%, while reducing training time by a factor of 10. Overall, our findings challenge the common belief that graph neural networks work equally well independent of dataset size and diversity, and suggest that caution must be exercised when making generalizations based on narrow datasets.
Rotation Invariant Graph Neural Networks using Spin Convolutions
Jun 17, 2021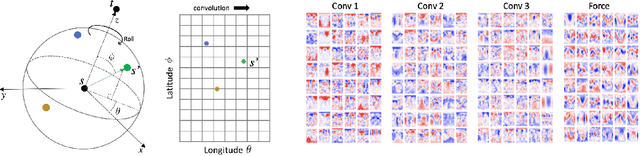
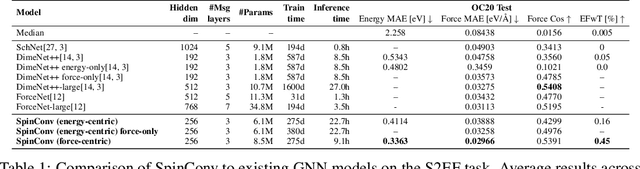
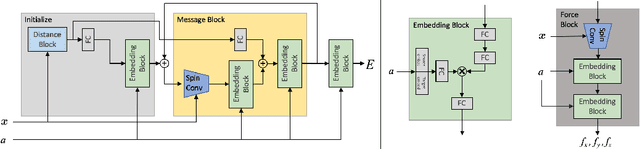
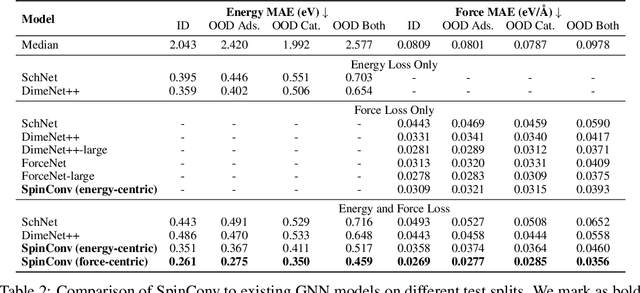
Progress towards the energy breakthroughs needed to combat climate change can be significantly accelerated through the efficient simulation of atomic systems. Simulation techniques based on first principles, such as Density Functional Theory (DFT), are limited in their practical use due to their high computational expense. Machine learning approaches have the potential to approximate DFT in a computationally efficient manner, which could dramatically increase the impact of computational simulations on real-world problems. Approximating DFT poses several challenges. These include accurately modeling the subtle changes in the relative positions and angles between atoms, and enforcing constraints such as rotation invariance or energy conservation. We introduce a novel approach to modeling angular information between sets of neighboring atoms in a graph neural network. Rotation invariance is achieved for the network's edge messages through the use of a per-edge local coordinate frame and a novel spin convolution over the remaining degree of freedom. Two model variants are proposed for the applications of structure relaxation and molecular dynamics. State-of-the-art results are demonstrated on the large-scale Open Catalyst 2020 dataset. Comparisons are also performed on the MD17 and QM9 datasets.
The Open Catalyst 2020 Dataset and Community Challenges
Oct 20, 2020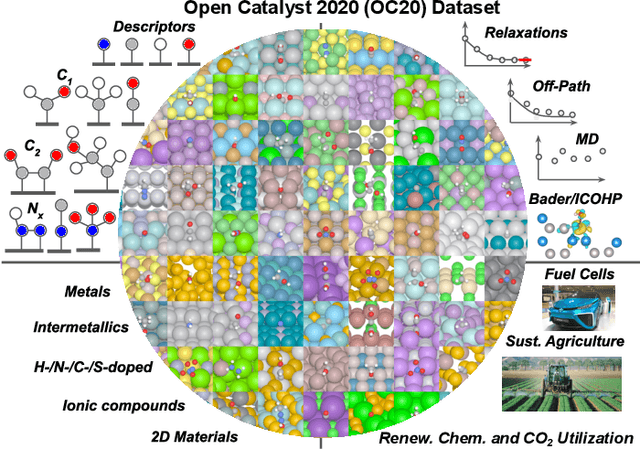

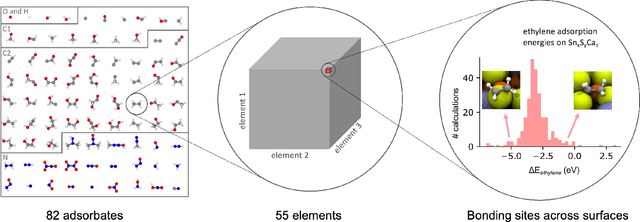
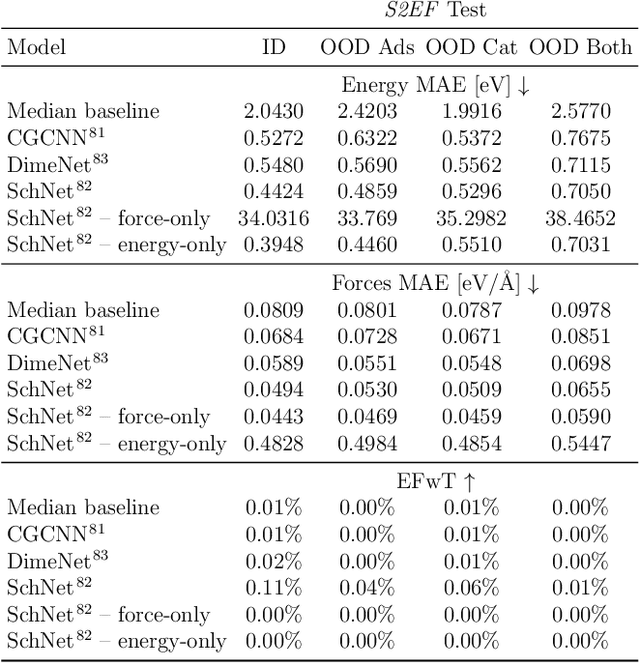
Catalyst discovery and optimization is key to solving many societal and energy challenges including solar fuels synthesis, long-term energy storage, and renewable fertilizer production. Despite considerable effort by the catalysis community to apply machine learning models to the computational catalyst discovery process, it remains an open challenge to build models that can generalize across both elemental compositions of surfaces and adsorbate identity/configurations, perhaps because datasets have been smaller in catalysis than related fields. To address this we developed the OC20 dataset, consisting of 1,281,121 Density Functional Theory (DFT) relaxations (264,900,500 single point evaluations) across a wide swath of materials, surfaces, and adsorbates (nitrogen, carbon, and oxygen chemistries). We supplemented this dataset with randomly perturbed structures, short timescale molecular dynamics, and electronic structure analyses. The dataset comprises three central tasks indicative of day-to-day catalyst modeling and comes with pre-defined train/validation/test splits to facilitate direct comparisons with future model development efforts. We applied three state-of-the-art graph neural network models (SchNet, Dimenet, CGCNN) to each of these tasks as baseline demonstrations for the community to build on. In almost every task, no upper limit on model size was identified, suggesting that even larger models are likely to improve on initial results. The dataset and baseline models are both provided as open resources, as well as a public leader board to encourage community contributions to solve these important tasks.
An Introduction to Electrocatalyst Design using Machine Learning for Renewable Energy Storage
Oct 14, 2020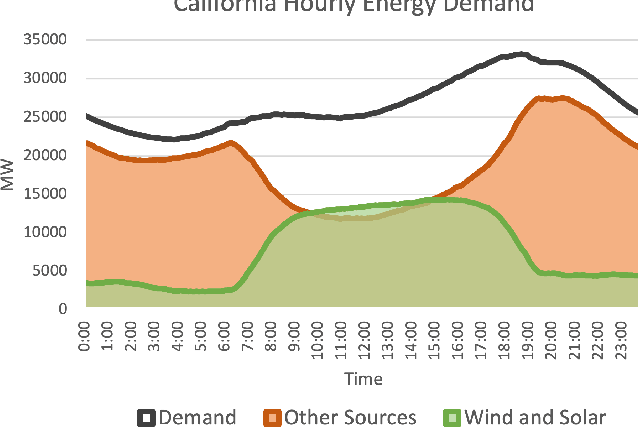
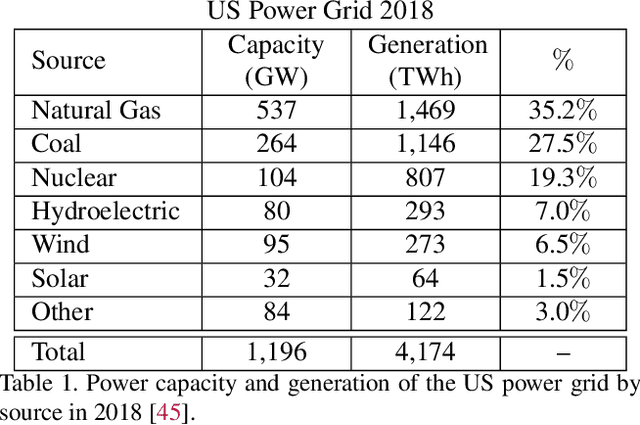
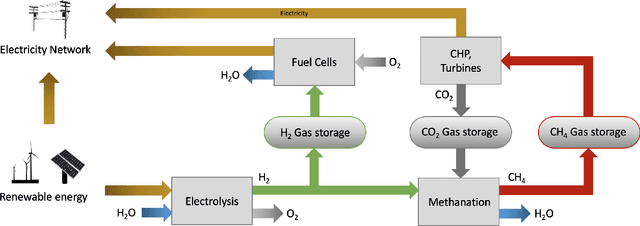
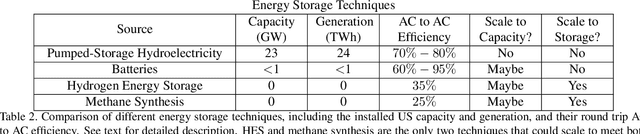
Scalable and cost-effective solutions to renewable energy storage are essential to addressing the world's rising energy needs while reducing climate change. As we increase our reliance on renewable energy sources such as wind and solar, which produce intermittent power, storage is needed to transfer power from times of peak generation to peak demand. This may require the storage of power for hours, days, or months. One solution that offers the potential of scaling to nation-sized grids is the conversion of renewable energy to other fuels, such as hydrogen or methane. To be widely adopted, this process requires cost-effective solutions to running electrochemical reactions. An open challenge is finding low-cost electrocatalysts to drive these reactions at high rates. Through the use of quantum mechanical simulations (density functional theory), new catalyst structures can be tested and evaluated. Unfortunately, the high computational cost of these simulations limits the number of structures that may be tested. The use of machine learning may provide a method to efficiently approximate these calculations, leading to new approaches in finding effective electrocatalysts. In this paper, we provide an introduction to the challenges in finding suitable electrocatalysts, how machine learning may be applied to the problem, and the use of the Open Catalyst Project OC20 dataset for model training.