 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRotation Invariant Graph Neural Networks using Spin Convolutions
Paper and Code
Jun 17, 2021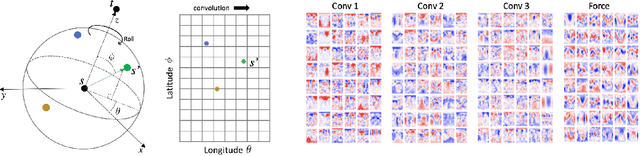
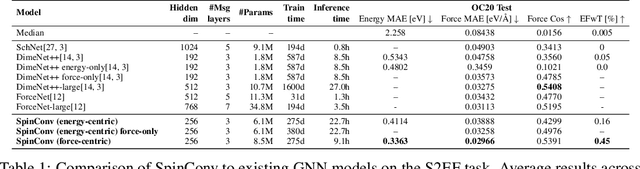
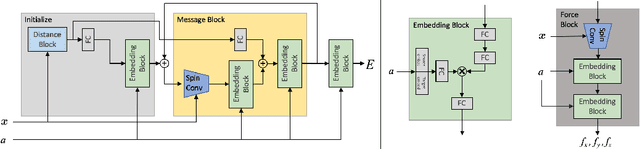
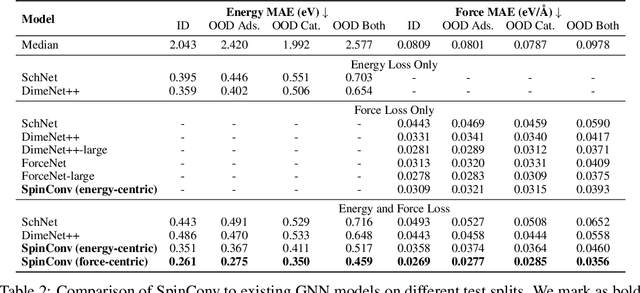
Progress towards the energy breakthroughs needed to combat climate change can be significantly accelerated through the efficient simulation of atomic systems. Simulation techniques based on first principles, such as Density Functional Theory (DFT), are limited in their practical use due to their high computational expense. Machine learning approaches have the potential to approximate DFT in a computationally efficient manner, which could dramatically increase the impact of computational simulations on real-world problems. Approximating DFT poses several challenges. These include accurately modeling the subtle changes in the relative positions and angles between atoms, and enforcing constraints such as rotation invariance or energy conservation. We introduce a novel approach to modeling angular information between sets of neighboring atoms in a graph neural network. Rotation invariance is achieved for the network's edge messages through the use of a per-edge local coordinate frame and a novel spin convolution over the remaining degree of freedom. Two model variants are proposed for the applications of structure relaxation and molecular dynamics. State-of-the-art results are demonstrated on the large-scale Open Catalyst 2020 dataset. Comparisons are also performed on the MD17 and QM9 datasets.