 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMATRIX: A Multimodal Benchmark and Post-Training Framework for Materials Science
Jan 30, 2026Scientific reasoning in materials science requires integrating multimodal experimental evidence with underlying physical theory. Existing benchmarks make it difficult to assess whether incorporating visual experimental data during post-training improves mechanism-grounded explanation reasoning beyond text-only supervision. We introduce MATRIX, a multimodal benchmark for materials science reasoning that evaluates foundational theory, research-level reasoning, and the interpretation of real experimental artifacts across multiple characterization modalities. Using MATRIX as a controlled diagnostic, we isolate the effect of visual grounding by comparing post-training on structured materials science text alone with post-training that incorporates paired experimental images. Despite using relatively small amounts of multimodal data, visual supervision improves experimental interpretation by 10-25% and yields 5-16% gains on text-only scientific reasoning tasks. Our results demonstrate that these improvements rely on correct image-text alignment during post-training, highlighting cross-modal representational transfer. We also observe consistent improvements on ScienceQA and PubMedQA, demonstrating that the benefits of structured multimodal post-training extend beyond materials science. The MATRIX dataset is available at https://huggingface.co/datasets/radical-ai/MATRIX and the model at https://huggingface.co/radical-ai/MATRIX-PT.
AdsorbDiff: Adsorbate Placement via Conditional Denoising Diffusion
May 07, 2024Determining the optimal configuration of adsorbates on a slab (adslab) is pivotal in the exploration of novel catalysts across diverse applications. Traditionally, the quest for the lowest energy adslab configuration involves placing the adsorbate onto the slab followed by an optimization process. Prior methodologies have relied on heuristics, problem-specific intuitions, or brute-force approaches to guide adsorbate placement. In this work, we propose a novel framework for adsorbate placement using denoising diffusion. The model is designed to predict the optimal adsorbate site and orientation corresponding to the lowest energy configuration. Further, we have an end-to-end evaluation framework where diffusion-predicted adslab configuration is optimized with a pretrained machine learning force field and finally evaluated with Density Functional Theory (DFT). Our findings demonstrate an acceleration of up to 5x or 3.5x improvement in accuracy compared to the previous best approach. Given the novelty of this framework and application, we provide insights into the impact of pre-training, model architectures, and conduct extensive experiments to underscore the significance of this approach.
From Molecules to Materials: Pre-training Large Generalizable Models for Atomic Property Prediction
Oct 25, 2023Foundation models have been transformational in machine learning fields such as natural language processing and computer vision. Similar success in atomic property prediction has been limited due to the challenges of training effective models across multiple chemical domains. To address this, we introduce Joint Multi-domain Pre-training (JMP), a supervised pre-training strategy that simultaneously trains on multiple datasets from different chemical domains, treating each dataset as a unique pre-training task within a multi-task framework. Our combined training dataset consists of $\sim$120M systems from OC20, OC22, ANI-1x, and Transition-1x. We evaluate performance and generalization by fine-tuning over a diverse set of downstream tasks and datasets including: QM9, rMD17, MatBench, QMOF, SPICE, and MD22. JMP demonstrates an average improvement of 59% over training from scratch, and matches or sets state-of-the-art on 34 out of 40 tasks. Our work highlights the potential of pre-training strategies that utilize diverse data to advance property prediction across chemical domains, especially for low-data tasks.
Spherical Channels for Modeling Atomic Interactions
Jun 29, 2022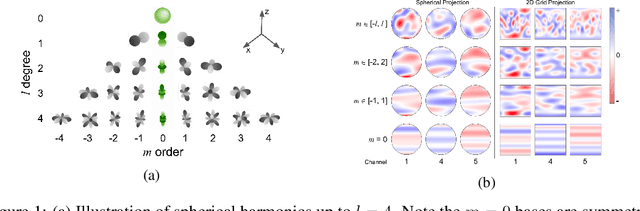
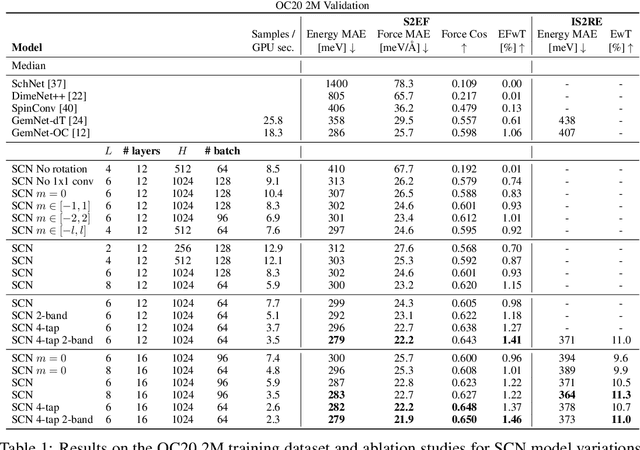
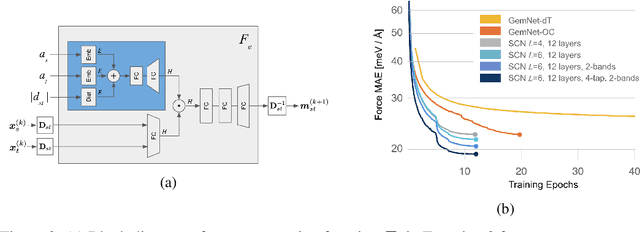
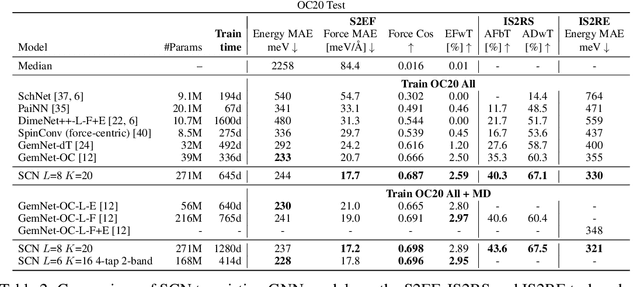
Modeling the energy and forces of atomic systems is a fundamental problem in computational chemistry with the potential to help address many of the world's most pressing problems, including those related to energy scarcity and climate change. These calculations are traditionally performed using Density Functional Theory, which is computationally very expensive. Machine learning has the potential to dramatically improve the efficiency of these calculations from days or hours to seconds. We propose the Spherical Channel Network (SCN) to model atomic energies and forces. The SCN is a graph neural network where nodes represent atoms and edges their neighboring atoms. The atom embeddings are a set of spherical functions, called spherical channels, represented using spherical harmonics. We demonstrate, that by rotating the embeddings based on the 3D edge orientation, more information may be utilized while maintaining the rotational equivariance of the messages. While equivariance is a desirable property, we find that by relaxing this constraint in both message passing and aggregation, improved accuracy may be achieved. We demonstrate state-of-the-art results on the large-scale Open Catalyst 2020 dataset in both energy and force prediction for numerous tasks and metrics.
The Open Catalyst 2022 Dataset and Challenges for Oxide Electrocatalysis
Jun 17, 2022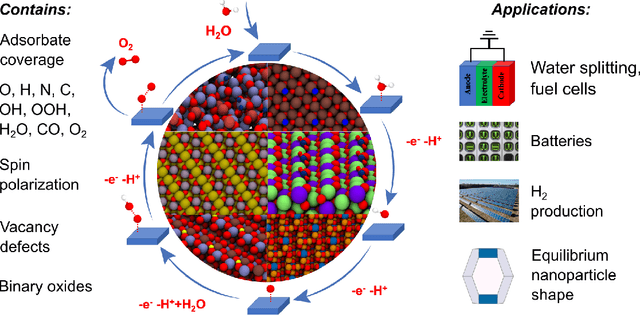
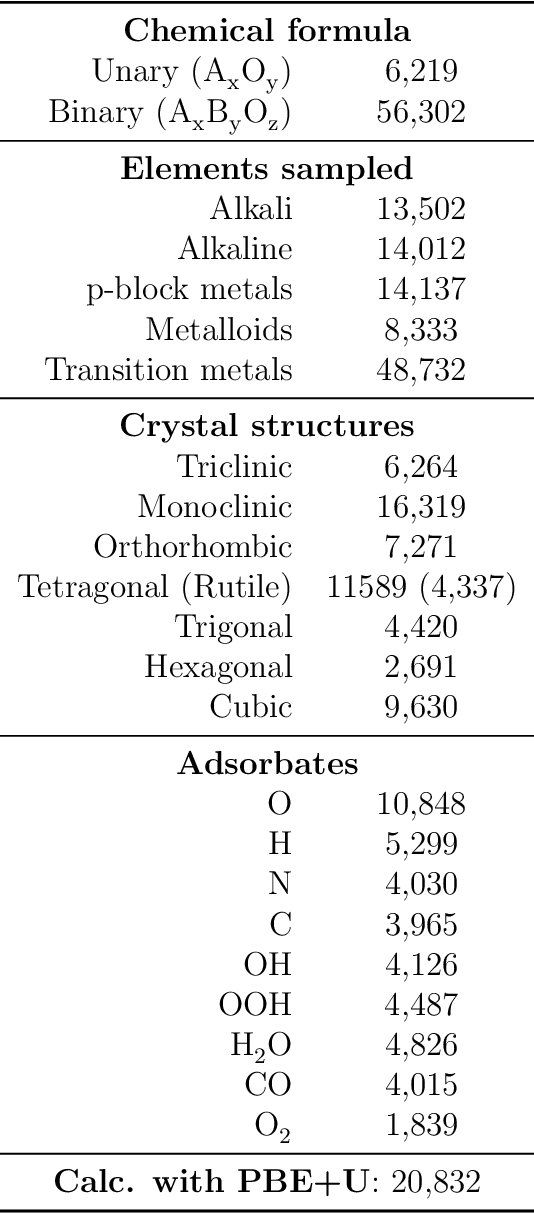
Computational catalysis and machine learning communities have made considerable progress in developing machine learning models for catalyst discovery and design. Yet, a general machine learning potential that spans the chemical space of catalysis is still out of reach. A significant hurdle is obtaining access to training data across a wide range of materials. One important class of materials where data is lacking are oxides, which inhibits models from studying the Oxygen Evolution Reaction and oxide electrocatalysis more generally. To address this we developed the Open Catalyst 2022(OC22) dataset, consisting of 62,521 Density Functional Theory (DFT) relaxations (~9,884,504 single point calculations) across a range of oxide materials, coverages, and adsorbates (*H, *O, *N, *C, *OOH, *OH, *OH2, *O2, *CO). We define generalized tasks to predict the total system energy that are applicable across catalysis, develop baseline performance of several graph neural networks (SchNet, DimeNet++, ForceNet, SpinConv, PaiNN, GemNet-dT, GemNet-OC), and provide pre-defined dataset splits to establish clear benchmarks for future efforts. For all tasks, we study whether combining datasets leads to better results, even if they contain different materials or adsorbates. Specifically, we jointly train models on Open Catalyst 2020 (OC20) Dataset and OC22, or fine-tune pretrained OC20 models on OC22. In the most general task, GemNet-OC sees a ~32% improvement in energy predictions through fine-tuning and a ~9% improvement in force predictions via joint training. Surprisingly, joint training on both the OC20 and much smaller OC22 datasets also improves total energy predictions on OC20 by ~19%. The dataset and baseline models are open sourced, and a public leaderboard will follow to encourage continued community developments on the total energy tasks and data.
Rotation Invariant Graph Neural Networks using Spin Convolutions
Jun 17, 2021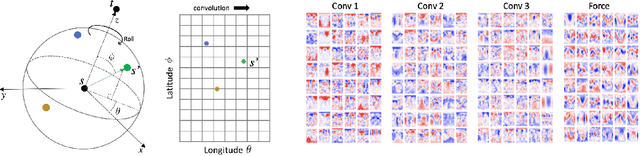
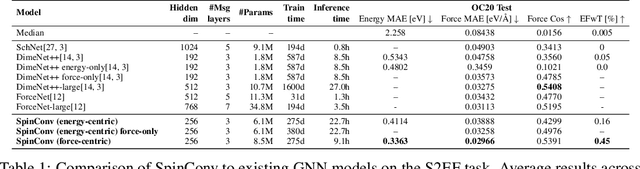
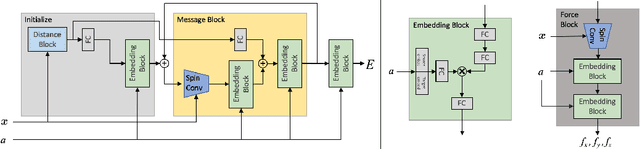
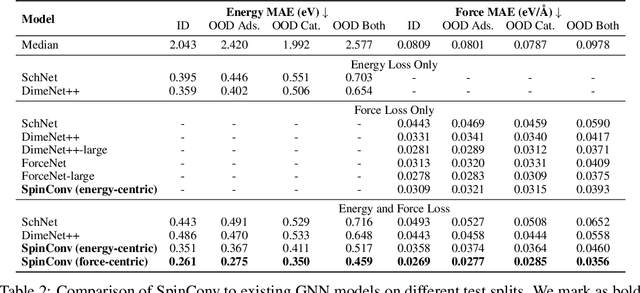
Progress towards the energy breakthroughs needed to combat climate change can be significantly accelerated through the efficient simulation of atomic systems. Simulation techniques based on first principles, such as Density Functional Theory (DFT), are limited in their practical use due to their high computational expense. Machine learning approaches have the potential to approximate DFT in a computationally efficient manner, which could dramatically increase the impact of computational simulations on real-world problems. Approximating DFT poses several challenges. These include accurately modeling the subtle changes in the relative positions and angles between atoms, and enforcing constraints such as rotation invariance or energy conservation. We introduce a novel approach to modeling angular information between sets of neighboring atoms in a graph neural network. Rotation invariance is achieved for the network's edge messages through the use of a per-edge local coordinate frame and a novel spin convolution over the remaining degree of freedom. Two model variants are proposed for the applications of structure relaxation and molecular dynamics. State-of-the-art results are demonstrated on the large-scale Open Catalyst 2020 dataset. Comparisons are also performed on the MD17 and QM9 datasets.