 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCausalVQA: A Physically Grounded Causal Reasoning Benchmark for Video Models
Jun 11, 2025We introduce CausalVQA, a benchmark dataset for video question answering (VQA) composed of question-answer pairs that probe models' understanding of causality in the physical world. Existing VQA benchmarks either tend to focus on surface perceptual understanding of real-world videos, or on narrow physical reasoning questions created using simulation environments. CausalVQA fills an important gap by presenting challenging questions that are grounded in real-world scenarios, while focusing on models' ability to predict the likely outcomes of different actions and events through five question types: counterfactual, hypothetical, anticipation, planning and descriptive. We designed quality control mechanisms that prevent models from exploiting trivial shortcuts, requiring models to base their answers on deep visual understanding instead of linguistic cues. We find that current frontier multimodal models fall substantially below human performance on the benchmark, especially on anticipation and hypothetical questions. This highlights a challenge for current systems to leverage spatial-temporal reasoning, understanding of physical principles, and comprehension of possible alternatives to make accurate predictions in real-world settings.
V-JEPA 2: Self-Supervised Video Models Enable Understanding, Prediction and Planning
Jun 11, 2025A major challenge for modern AI is to learn to understand the world and learn to act largely by observation. This paper explores a self-supervised approach that combines internet-scale video data with a small amount of interaction data (robot trajectories), to develop models capable of understanding, predicting, and planning in the physical world. We first pre-train an action-free joint-embedding-predictive architecture, V-JEPA 2, on a video and image dataset comprising over 1 million hours of internet video. V-JEPA 2 achieves strong performance on motion understanding (77.3 top-1 accuracy on Something-Something v2) and state-of-the-art performance on human action anticipation (39.7 recall-at-5 on Epic-Kitchens-100) surpassing previous task-specific models. Additionally, after aligning V-JEPA 2 with a large language model, we demonstrate state-of-the-art performance on multiple video question-answering tasks at the 8 billion parameter scale (e.g., 84.0 on PerceptionTest, 76.9 on TempCompass). Finally, we show how self-supervised learning can be applied to robotic planning tasks by post-training a latent action-conditioned world model, V-JEPA 2-AC, using less than 62 hours of unlabeled robot videos from the Droid dataset. We deploy V-JEPA 2-AC zero-shot on Franka arms in two different labs and enable picking and placing of objects using planning with image goals. Notably, this is achieved without collecting any data from the robots in these environments, and without any task-specific training or reward. This work demonstrates how self-supervised learning from web-scale data and a small amount of robot interaction data can yield a world model capable of planning in the physical world.
Open Materials 2024 (OMat24) Inorganic Materials Dataset and Models
Oct 16, 2024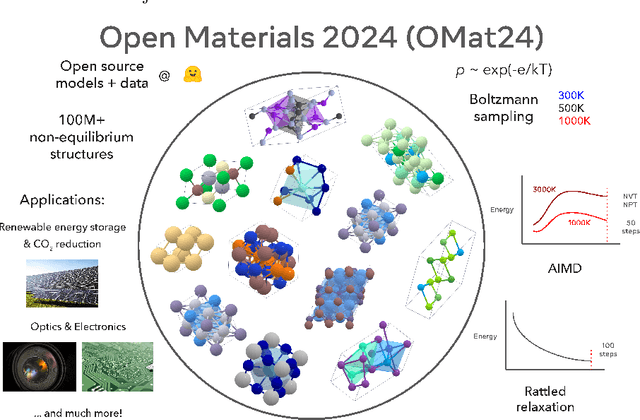
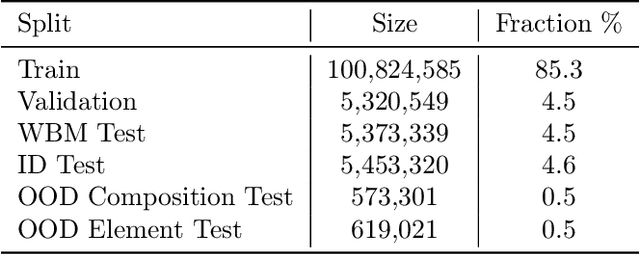
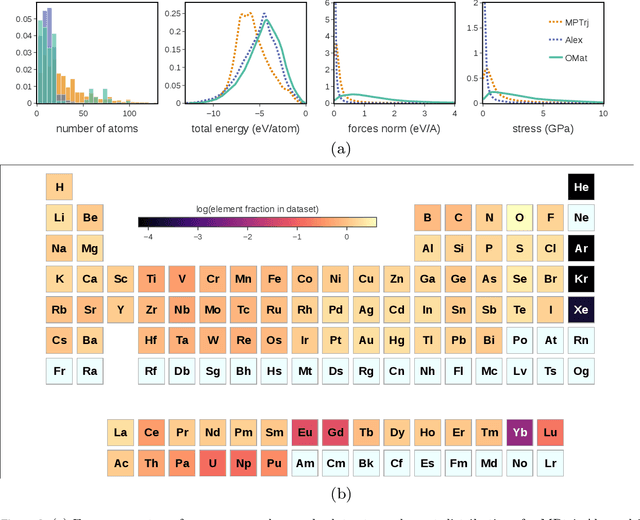
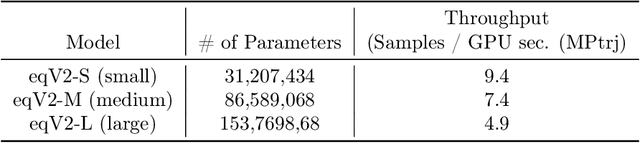
The ability to discover new materials with desirable properties is critical for numerous applications from helping mitigate climate change to advances in next generation computing hardware. AI has the potential to accelerate materials discovery and design by more effectively exploring the chemical space compared to other computational methods or by trial-and-error. While substantial progress has been made on AI for materials data, benchmarks, and models, a barrier that has emerged is the lack of publicly available training data and open pre-trained models. To address this, we present a Meta FAIR release of the Open Materials 2024 (OMat24) large-scale open dataset and an accompanying set of pre-trained models. OMat24 contains over 110 million density functional theory (DFT) calculations focused on structural and compositional diversity. Our EquiformerV2 models achieve state-of-the-art performance on the Matbench Discovery leaderboard and are capable of predicting ground-state stability and formation energies to an F1 score above 0.9 and an accuracy of 20 meV/atom, respectively. We explore the impact of model size, auxiliary denoising objectives, and fine-tuning on performance across a range of datasets including OMat24, MPtraj, and Alexandria. The open release of the OMat24 dataset and models enables the research community to build upon our efforts and drive further advancements in AI-assisted materials science.
The Open Catalyst 2022 Dataset and Challenges for Oxide Electrocatalysis
Jun 17, 2022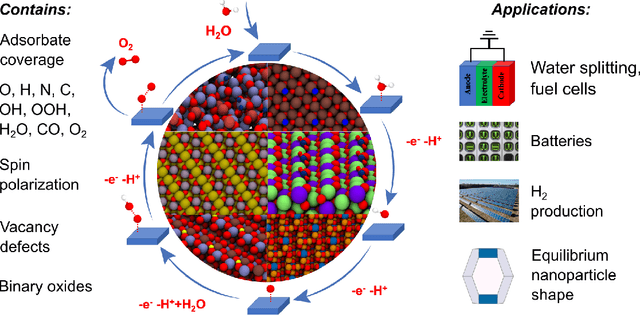
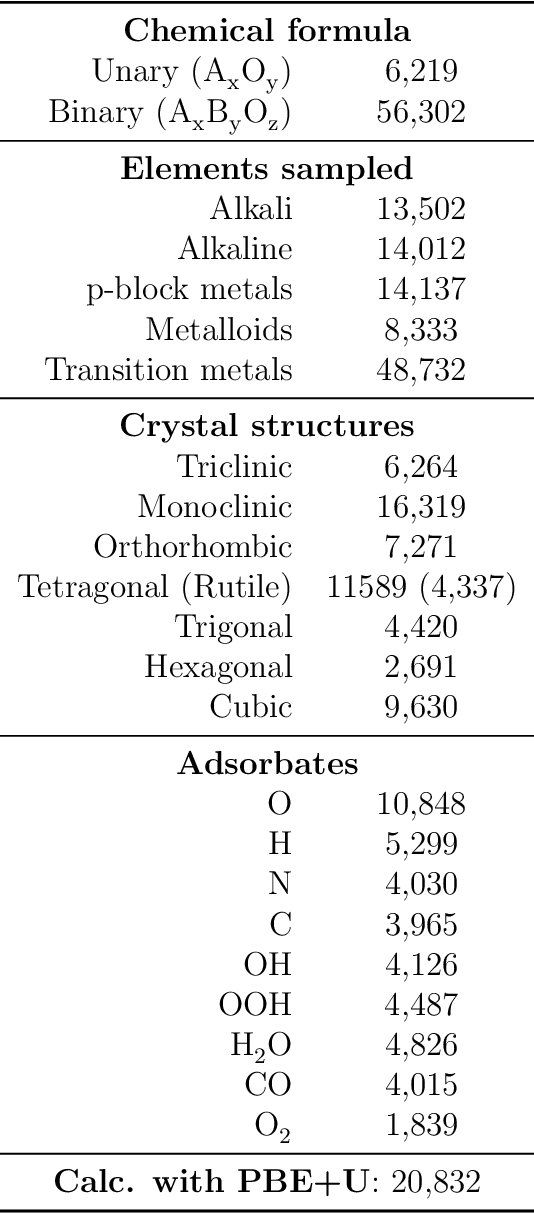
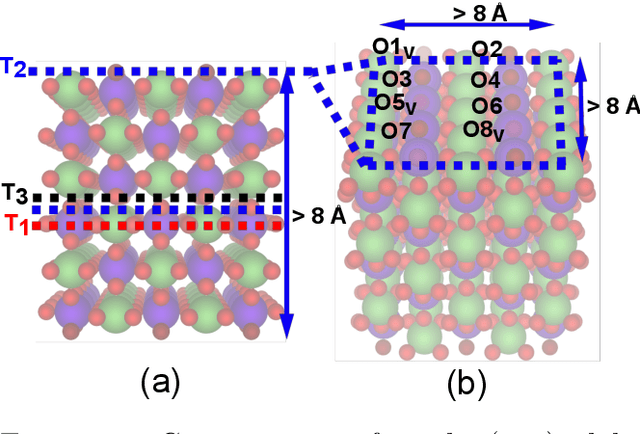
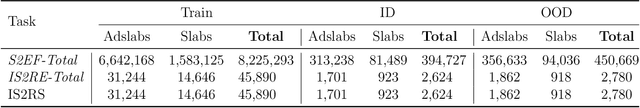
Computational catalysis and machine learning communities have made considerable progress in developing machine learning models for catalyst discovery and design. Yet, a general machine learning potential that spans the chemical space of catalysis is still out of reach. A significant hurdle is obtaining access to training data across a wide range of materials. One important class of materials where data is lacking are oxides, which inhibits models from studying the Oxygen Evolution Reaction and oxide electrocatalysis more generally. To address this we developed the Open Catalyst 2022(OC22) dataset, consisting of 62,521 Density Functional Theory (DFT) relaxations (~9,884,504 single point calculations) across a range of oxide materials, coverages, and adsorbates (*H, *O, *N, *C, *OOH, *OH, *OH2, *O2, *CO). We define generalized tasks to predict the total system energy that are applicable across catalysis, develop baseline performance of several graph neural networks (SchNet, DimeNet++, ForceNet, SpinConv, PaiNN, GemNet-dT, GemNet-OC), and provide pre-defined dataset splits to establish clear benchmarks for future efforts. For all tasks, we study whether combining datasets leads to better results, even if they contain different materials or adsorbates. Specifically, we jointly train models on Open Catalyst 2020 (OC20) Dataset and OC22, or fine-tune pretrained OC20 models on OC22. In the most general task, GemNet-OC sees a ~32% improvement in energy predictions through fine-tuning and a ~9% improvement in force predictions via joint training. Surprisingly, joint training on both the OC20 and much smaller OC22 datasets also improves total energy predictions on OC20 by ~19%. The dataset and baseline models are open sourced, and a public leaderboard will follow to encourage continued community developments on the total energy tasks and data.