 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAll-atom Diffusion Transformers: Unified generative modelling of molecules and materials
Mar 05, 2025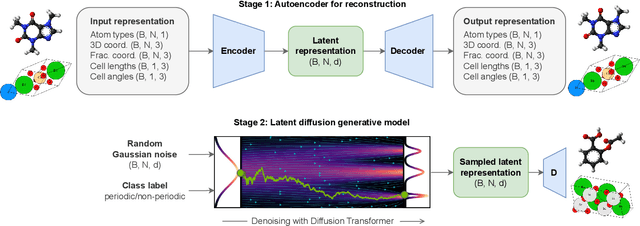

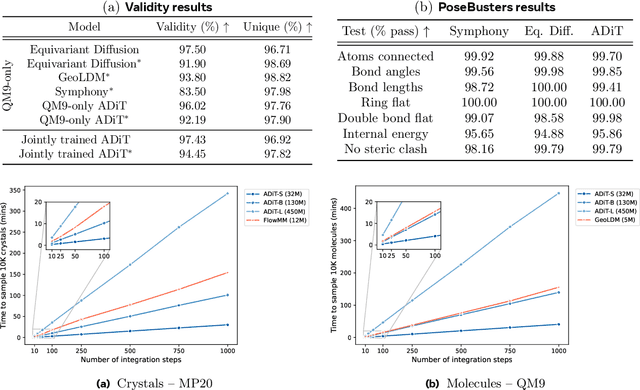
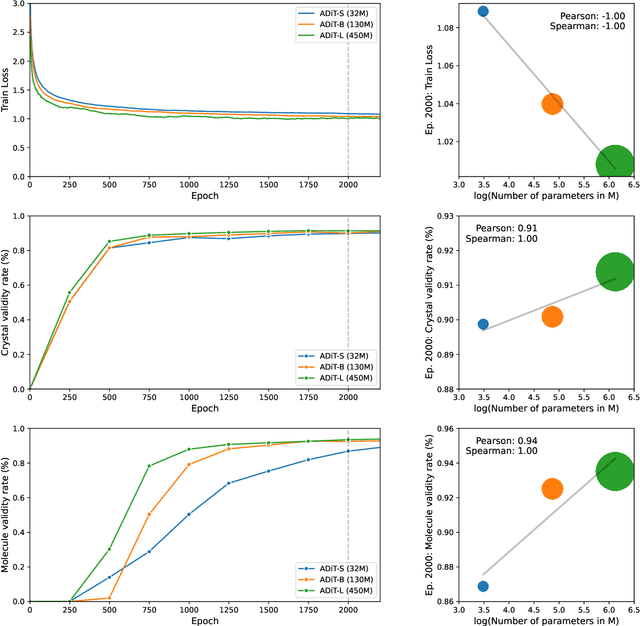
Diffusion models are the standard toolkit for generative modelling of 3D atomic systems. However, for different types of atomic systems - such as molecules and materials - the generative processes are usually highly specific to the target system despite the underlying physics being the same. We introduce the All-atom Diffusion Transformer (ADiT), a unified latent diffusion framework for jointly generating both periodic materials and non-periodic molecular systems using the same model: (1) An autoencoder maps a unified, all-atom representations of molecules and materials to a shared latent embedding space; and (2) A diffusion model is trained to generate new latent embeddings that the autoencoder can decode to sample new molecules or materials. Experiments on QM9 and MP20 datasets demonstrate that jointly trained ADiT generates realistic and valid molecules as well as materials, exceeding state-of-the-art results from molecule and crystal-specific models. ADiT uses standard Transformers for both the autoencoder and diffusion model, resulting in significant speedups during training and inference compared to equivariant diffusion models. Scaling ADiT up to half a billion parameters predictably improves performance, representing a step towards broadly generalizable foundation models for generative chemistry. Open source code: https://github.com/facebookresearch/all-atom-diffusion-transformer
Open Materials 2024 (OMat24) Inorganic Materials Dataset and Models
Oct 16, 2024
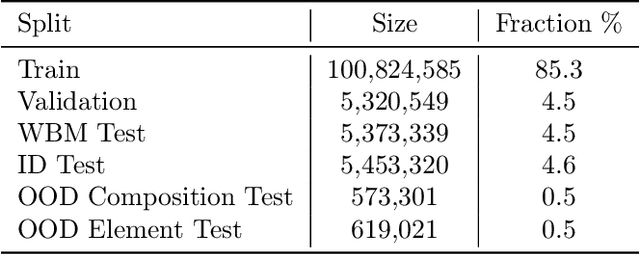
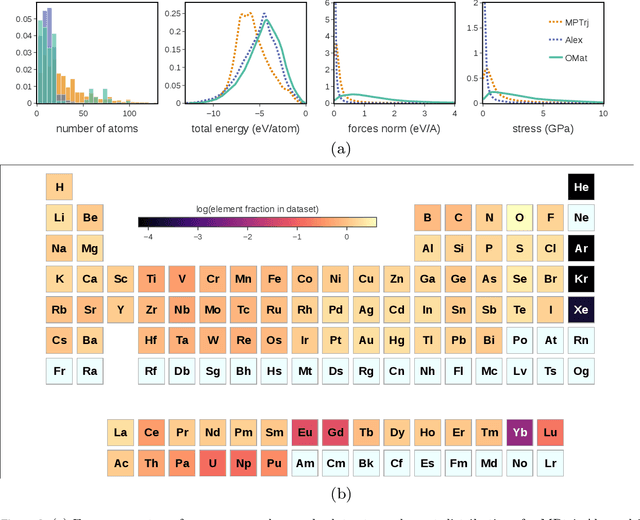
The ability to discover new materials with desirable properties is critical for numerous applications from helping mitigate climate change to advances in next generation computing hardware. AI has the potential to accelerate materials discovery and design by more effectively exploring the chemical space compared to other computational methods or by trial-and-error. While substantial progress has been made on AI for materials data, benchmarks, and models, a barrier that has emerged is the lack of publicly available training data and open pre-trained models. To address this, we present a Meta FAIR release of the Open Materials 2024 (OMat24) large-scale open dataset and an accompanying set of pre-trained models. OMat24 contains over 110 million density functional theory (DFT) calculations focused on structural and compositional diversity. Our EquiformerV2 models achieve state-of-the-art performance on the Matbench Discovery leaderboard and are capable of predicting ground-state stability and formation energies to an F1 score above 0.9 and an accuracy of 20 meV/atom, respectively. We explore the impact of model size, auxiliary denoising objectives, and fine-tuning on performance across a range of datasets including OMat24, MPtraj, and Alexandria. The open release of the OMat24 dataset and models enables the research community to build upon our efforts and drive further advancements in AI-assisted materials science.
Adapting OC20-trained EquiformerV2 Models for High-Entropy Materials
Mar 14, 2024
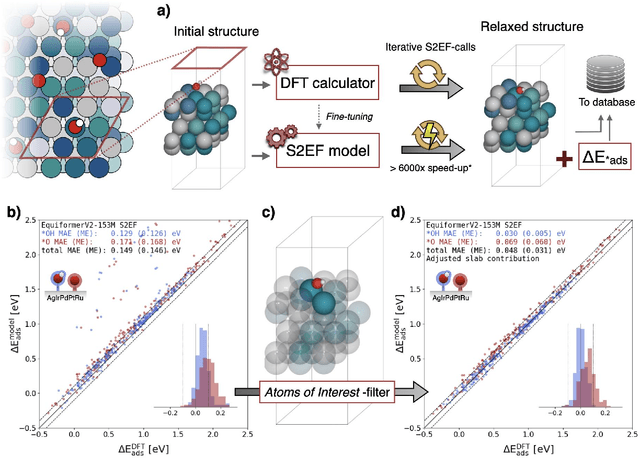

Computational high-throughput studies, especially in research on high-entropy materials and catalysts, are hampered by high-dimensional composition spaces and myriad structural microstates. They present bottlenecks to the conventional use of density functional theory calculations, and consequently, the use of machine-learned potentials is becoming increasingly prevalent in atomic structure simulations. In this communication, we show the results of adjusting and fine-tuning the pretrained EquiformerV2 model from the Open Catalyst Project to infer adsorption energies of *OH and *O on the out-of-domain high-entropy alloy Ag-Ir-Pd-Pt-Ru. By applying an energy filter based on the local environment of the binding site the zero-shot inference is markedly improved and through few-shot fine-tuning the model yields state-of-the-art accuracy. It is also found that EquiformerV2, assuming the role of general machine learning potential, is able to inform a smaller, more focused direct inference model. This knowledge distillation setup boosts performance on complex binding sites. Collectively, this shows that foundational knowledge learned from ordered intermetallic structures, can be extrapolated to the highly disordered structures of solid-solutions. With the vastly accelerated computational throughput of these models, hitherto infeasible research in the high-entropy material space is now readily accessible.
From Molecules to Materials: Pre-training Large Generalizable Models for Atomic Property Prediction
Oct 25, 2023Foundation models have been transformational in machine learning fields such as natural language processing and computer vision. Similar success in atomic property prediction has been limited due to the challenges of training effective models across multiple chemical domains. To address this, we introduce Joint Multi-domain Pre-training (JMP), a supervised pre-training strategy that simultaneously trains on multiple datasets from different chemical domains, treating each dataset as a unique pre-training task within a multi-task framework. Our combined training dataset consists of $\sim$120M systems from OC20, OC22, ANI-1x, and Transition-1x. We evaluate performance and generalization by fine-tuning over a diverse set of downstream tasks and datasets including: QM9, rMD17, MatBench, QMOF, SPICE, and MD22. JMP demonstrates an average improvement of 59% over training from scratch, and matches or sets state-of-the-art on 34 out of 40 tasks. Our work highlights the potential of pre-training strategies that utilize diverse data to advance property prediction across chemical domains, especially for low-data tasks.
AdsorbML: Accelerating Adsorption Energy Calculations with Machine Learning
Nov 29, 2022Computational catalysis is playing an increasingly significant role in the design of catalysts across a wide range of applications. A common task for many computational methods is the need to accurately compute the minimum binding energy - the adsorption energy - for an adsorbate and a catalyst surface of interest. Traditionally, the identification of low energy adsorbate-surface configurations relies on heuristic methods and researcher intuition. As the desire to perform high-throughput screening increases, it becomes challenging to use heuristics and intuition alone. In this paper, we demonstrate machine learning potentials can be leveraged to identify low energy adsorbate-surface configurations more accurately and efficiently. Our algorithm provides a spectrum of trade-offs between accuracy and efficiency, with one balanced option finding the lowest energy configuration, within a 0.1 eV threshold, 86.63% of the time, while achieving a 1387x speedup in computation. To standardize benchmarking, we introduce the Open Catalyst Dense dataset containing nearly 1,000 diverse surfaces and 87,045 unique configurations.