 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBridging Offline and Online Reinforcement Learning for LLMs
Jun 26, 2025We investigate the effectiveness of reinforcement learning methods for finetuning large language models when transitioning from offline to semi-online to fully online regimes for both verifiable and non-verifiable tasks. Our experiments cover training on verifiable math as well as non-verifiable instruction following with a set of benchmark evaluations for both. Across these settings, we extensively compare online and semi-online Direct Preference Optimization and Group Reward Policy Optimization objectives, and surprisingly find similar performance and convergence between these variants, which all strongly outperform offline methods. We provide a detailed analysis of the training dynamics and hyperparameter selection strategies to achieve optimal results. Finally, we show that multi-tasking with verifiable and non-verifiable rewards jointly yields improved performance across both task types.
LLM Pretraining with Continuous Concepts
Feb 12, 2025



Next token prediction has been the standard training objective used in large language model pretraining. Representations are learned as a result of optimizing for token-level perplexity. We propose Continuous Concept Mixing (CoCoMix), a novel pretraining framework that combines discrete next token prediction with continuous concepts. Specifically, CoCoMix predicts continuous concepts learned from a pretrained sparse autoencoder and mixes them into the model's hidden state by interleaving with token hidden representations. Through experiments on multiple benchmarks, including language modeling and downstream reasoning tasks, we show that CoCoMix is more sample efficient and consistently outperforms standard next token prediction, knowledge distillation and inserting pause tokens. We find that combining both concept learning and interleaving in an end-to-end framework is critical to performance gains. Furthermore, CoCoMix enhances interpretability and steerability by allowing direct inspection and modification of the predicted concept, offering a transparent way to guide the model's internal reasoning process.
Thinking LLMs: General Instruction Following with Thought Generation
Oct 14, 2024



LLMs are typically trained to answer user questions or follow instructions similarly to how human experts respond. However, in the standard alignment framework they lack the basic ability of explicit thinking before answering. Thinking is important for complex questions that require reasoning and planning -- but can be applied to any task. We propose a training method for equipping existing LLMs with such thinking abilities for general instruction following without use of additional human data. We achieve this by an iterative search and optimization procedure that explores the space of possible thought generations, allowing the model to learn how to think without direct supervision. For each instruction, the thought candidates are scored using a judge model to evaluate their responses only, and then optimized via preference optimization. We show that this procedure leads to superior performance on AlpacaEval and Arena-Hard, and shows gains from thinking on non-reasoning categories such as marketing, health and general knowledge, in addition to more traditional reasoning & problem-solving tasks.
Rotationally Invariant Latent Distances for Uncertainty Estimation of Relaxed Energy Predictions by Graph Neural Network Potentials
Jul 15, 2024Graph neural networks (GNNs) have been shown to be astonishingly capable models for molecular property prediction, particularly as surrogates for expensive density functional theory calculations of relaxed energy for novel material discovery. However, one limitation of GNNs in this context is the lack of useful uncertainty prediction methods, as this is critical to the material discovery pipeline. In this work, we show that uncertainty quantification for relaxed energy calculations is more complex than uncertainty quantification for other kinds of molecular property prediction, due to the effect that structure optimizations have on the error distribution. We propose that distribution-free techniques are more useful tools for assessing calibration, recalibrating, and developing uncertainty prediction methods for GNNs performing relaxed energy calculations. We also develop a relaxed energy task for evaluating uncertainty methods for equivariant GNNs, based on distribution-free recalibration and using the Open Catalyst Project dataset. We benchmark a set of popular uncertainty prediction methods on this task, and show that latent distance methods, with our novel improvements, are the most well-calibrated and economical approach for relaxed energy calculations. Finally, we demonstrate that our latent space distance method produces results which align with our expectations on a clustering example, and on specific equation of state and adsorbate coverage examples from outside the training dataset.
AdsorbML: Accelerating Adsorption Energy Calculations with Machine Learning
Nov 29, 2022Computational catalysis is playing an increasingly significant role in the design of catalysts across a wide range of applications. A common task for many computational methods is the need to accurately compute the minimum binding energy - the adsorption energy - for an adsorbate and a catalyst surface of interest. Traditionally, the identification of low energy adsorbate-surface configurations relies on heuristic methods and researcher intuition. As the desire to perform high-throughput screening increases, it becomes challenging to use heuristics and intuition alone. In this paper, we demonstrate machine learning potentials can be leveraged to identify low energy adsorbate-surface configurations more accurately and efficiently. Our algorithm provides a spectrum of trade-offs between accuracy and efficiency, with one balanced option finding the lowest energy configuration, within a 0.1 eV threshold, 86.63% of the time, while achieving a 1387x speedup in computation. To standardize benchmarking, we introduce the Open Catalyst Dense dataset containing nearly 1,000 diverse surfaces and 87,045 unique configurations.
Spherical Channels for Modeling Atomic Interactions
Jun 29, 2022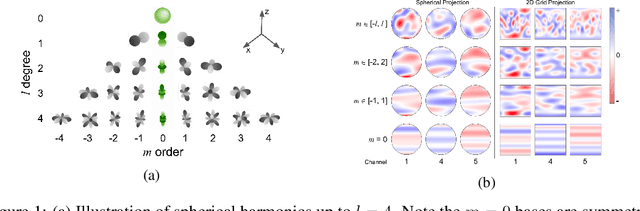
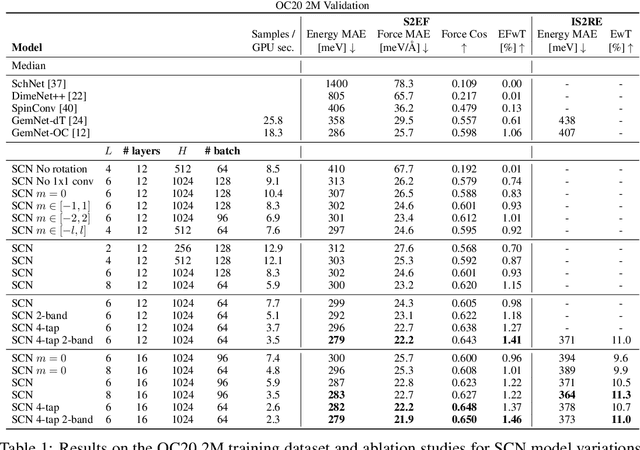
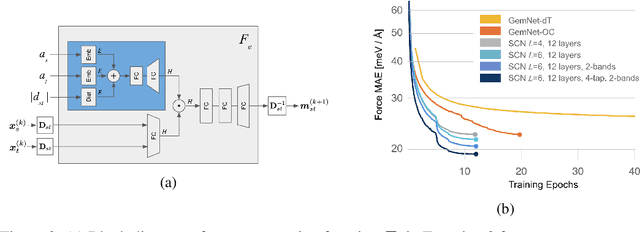
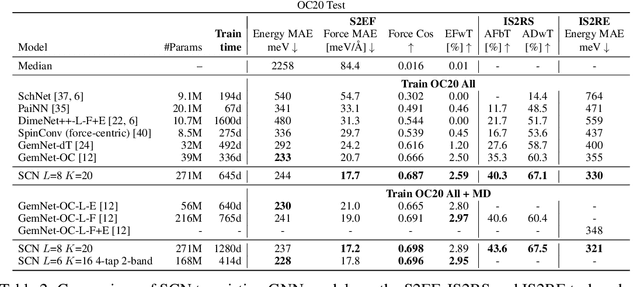
Modeling the energy and forces of atomic systems is a fundamental problem in computational chemistry with the potential to help address many of the world's most pressing problems, including those related to energy scarcity and climate change. These calculations are traditionally performed using Density Functional Theory, which is computationally very expensive. Machine learning has the potential to dramatically improve the efficiency of these calculations from days or hours to seconds. We propose the Spherical Channel Network (SCN) to model atomic energies and forces. The SCN is a graph neural network where nodes represent atoms and edges their neighboring atoms. The atom embeddings are a set of spherical functions, called spherical channels, represented using spherical harmonics. We demonstrate, that by rotating the embeddings based on the 3D edge orientation, more information may be utilized while maintaining the rotational equivariance of the messages. While equivariance is a desirable property, we find that by relaxing this constraint in both message passing and aggregation, improved accuracy may be achieved. We demonstrate state-of-the-art results on the large-scale Open Catalyst 2020 dataset in both energy and force prediction for numerous tasks and metrics.
The Open Catalyst 2022 Dataset and Challenges for Oxide Electrocatalysis
Jun 17, 2022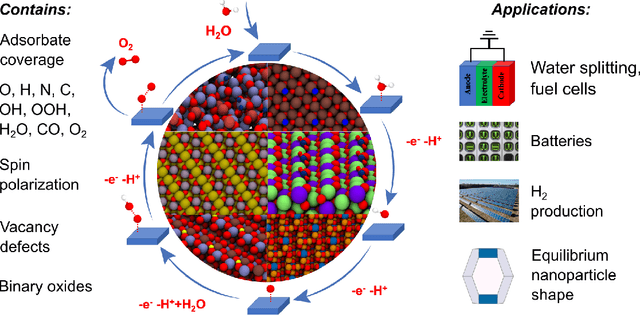
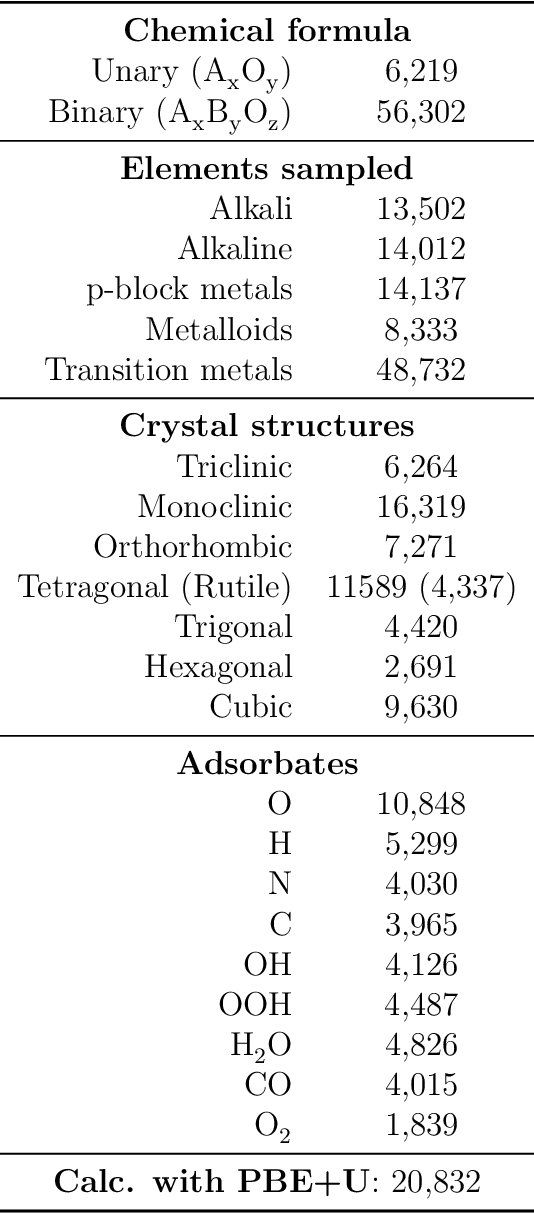
Computational catalysis and machine learning communities have made considerable progress in developing machine learning models for catalyst discovery and design. Yet, a general machine learning potential that spans the chemical space of catalysis is still out of reach. A significant hurdle is obtaining access to training data across a wide range of materials. One important class of materials where data is lacking are oxides, which inhibits models from studying the Oxygen Evolution Reaction and oxide electrocatalysis more generally. To address this we developed the Open Catalyst 2022(OC22) dataset, consisting of 62,521 Density Functional Theory (DFT) relaxations (~9,884,504 single point calculations) across a range of oxide materials, coverages, and adsorbates (*H, *O, *N, *C, *OOH, *OH, *OH2, *O2, *CO). We define generalized tasks to predict the total system energy that are applicable across catalysis, develop baseline performance of several graph neural networks (SchNet, DimeNet++, ForceNet, SpinConv, PaiNN, GemNet-dT, GemNet-OC), and provide pre-defined dataset splits to establish clear benchmarks for future efforts. For all tasks, we study whether combining datasets leads to better results, even if they contain different materials or adsorbates. Specifically, we jointly train models on Open Catalyst 2020 (OC20) Dataset and OC22, or fine-tune pretrained OC20 models on OC22. In the most general task, GemNet-OC sees a ~32% improvement in energy predictions through fine-tuning and a ~9% improvement in force predictions via joint training. Surprisingly, joint training on both the OC20 and much smaller OC22 datasets also improves total energy predictions on OC20 by ~19%. The dataset and baseline models are open sourced, and a public leaderboard will follow to encourage continued community developments on the total energy tasks and data.
Plug and Play Language Models: A Simple Approach to Controlled Text Generation
Jan 08, 2020
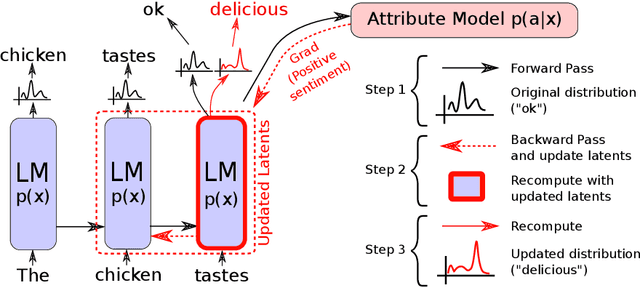
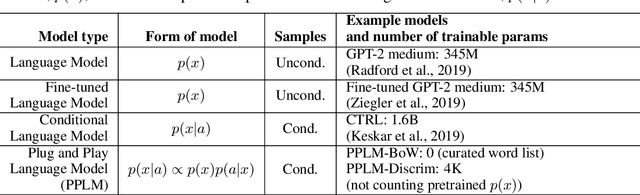
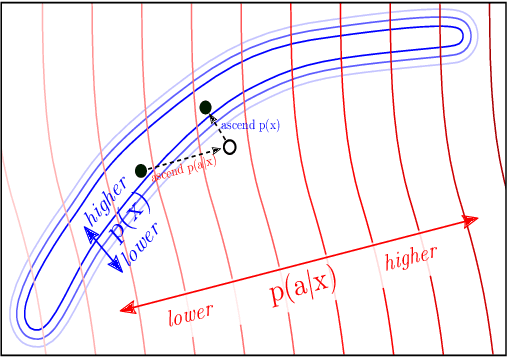
Large transformer-based language models (LMs) trained on huge text corpora have shown unparalleled generation capabilities. However, controlling attributes of the generated language (e.g. switching topic or sentiment) is difficult without modifying the model architecture or fine-tuning on attribute-specific data and entailing the significant cost of retraining. We propose a simple alternative: the Plug and Play Language Model (PPLM) for controllable language generation, which combines a pretrained LM with one or more simple attribute classifiers that guide text generation without any further training of the LM. In the canonical scenario we present, the attribute models are simple classifiers consisting of a user-specified bag of words or a single learned layer with 100,000 times fewer parameters than the LM. Sampling entails a forward and backward pass in which gradients from the attribute model push the LM's hidden activations and thus guide the generation. Model samples demonstrate control over a range of topics and sentiment styles, and extensive automated and human annotated evaluations show attribute alignment and fluency. PPLMs are flexible in that any combination of differentiable attribute models may be used to steer text generation, which will allow for diverse and creative applications beyond the examples given in this paper.
First-Order Preconditioning via Hypergradient Descent
Oct 18, 2019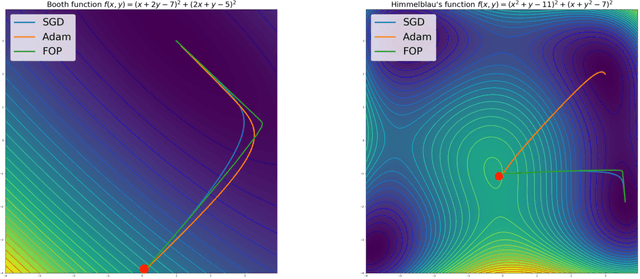
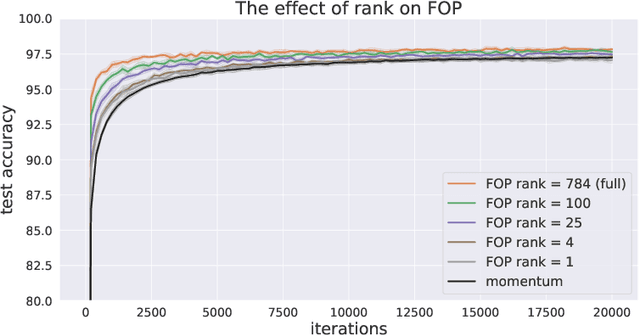
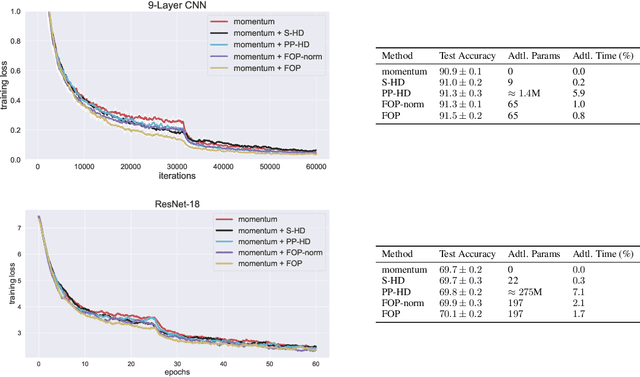
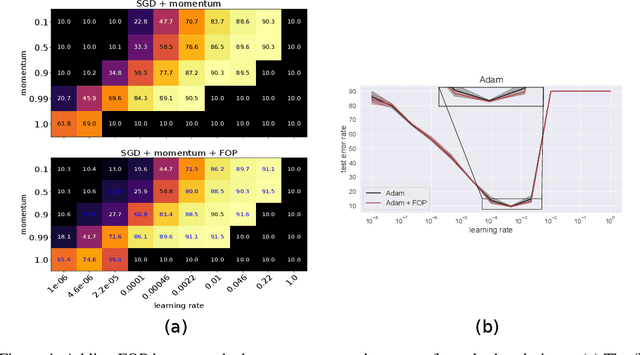
Standard gradient descent methods are susceptible to a range of issues that can impede training, such as high correlations and different scaling in parameter space. These difficulties can be addressed by second-order approaches that apply a preconditioning matrix to the gradient to improve convergence. Unfortunately, such algorithms typically struggle to scale to high-dimensional problems, in part because the calculation of specific preconditioners such as the inverse Hessian or Fisher information matrix is highly expensive. We introduce first-order preconditioning (FOP), a fast, scalable approach that generalizes previous work on hypergradient descent (Almeida et al., 1998; Maclaurin et al., 2015; Baydin et al., 2017) to learn a preconditioning matrix that only makes use of first-order information. Experiments show that FOP is able to improve the performance of standard deep learning optimizers on several visual classification tasks with minimal computational overhead. We also investigate the properties of the learned preconditioning matrices and perform a preliminary theoretical analysis of the algorithm.
LCA: Loss Change Allocation for Neural Network Training
Sep 03, 2019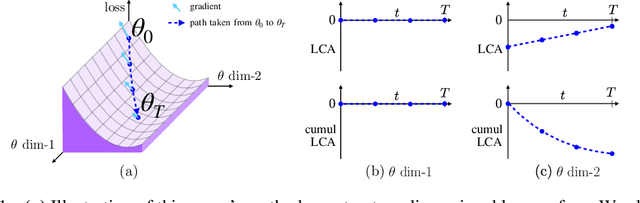

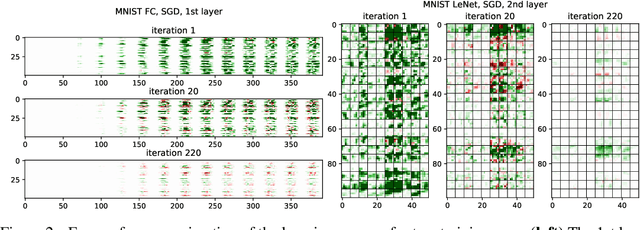
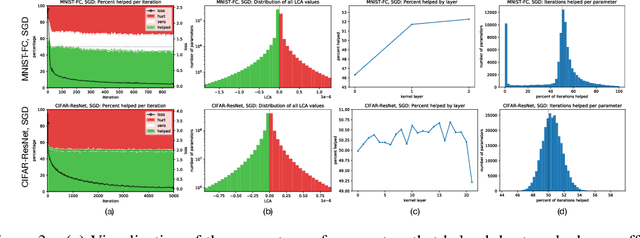
Neural networks enjoy widespread use, but many aspects of their training, representation, and operation are poorly understood. In particular, our view into the training process is limited, with a single scalar loss being the most common viewport into this high-dimensional, dynamic process. We propose a new window into training called Loss Change Allocation (LCA), in which credit for changes to the network loss is conservatively partitioned to the parameters. This measurement is accomplished by decomposing the components of an approximate path integral along the training trajectory using a Runge-Kutta integrator. This rich view shows which parameters are responsible for decreasing or increasing the loss during training, or which parameters "help" or "hurt" the network's learning, respectively. LCA may be summed over training iterations and/or over neurons, channels, or layers for increasingly coarse views. This new measurement device produces several insights into training. (1) We find that barely over 50% of parameters help during any given iteration. (2) Some entire layers hurt overall, moving on average against the training gradient, a phenomenon we hypothesize may be due to phase lag in an oscillatory training process. (3) Finally, increments in learning proceed in a synchronized manner across layers, often peaking on identical iterations.