 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAdjoint Sampling: Highly Scalable Diffusion Samplers via Adjoint Matching
Apr 16, 2025We introduce Adjoint Sampling, a highly scalable and efficient algorithm for learning diffusion processes that sample from unnormalized densities, or energy functions. It is the first on-policy approach that allows significantly more gradient updates than the number of energy evaluations and model samples, allowing us to scale to much larger problem settings than previously explored by similar methods. Our framework is theoretically grounded in stochastic optimal control and shares the same theoretical guarantees as Adjoint Matching, being able to train without the need for corrective measures that push samples towards the target distribution. We show how to incorporate key symmetries, as well as periodic boundary conditions, for modeling molecules in both cartesian and torsional coordinates. We demonstrate the effectiveness of our approach through extensive experiments on classical energy functions, and further scale up to neural network-based energy models where we perform amortized conformer generation across many molecular systems. To encourage further research in developing highly scalable sampling methods, we plan to open source these challenging benchmarks, where successful methods can directly impact progress in computational chemistry.
EquiformerV2: Improved Equivariant Transformer for Scaling to Higher-Degree Representations
Jun 21, 2023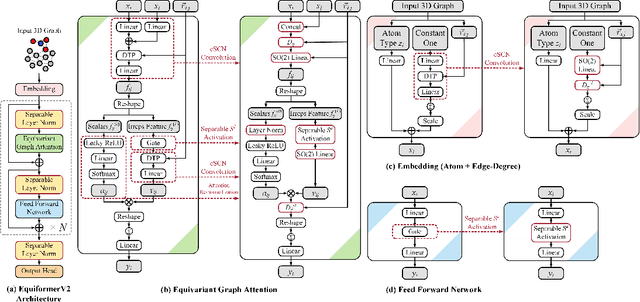
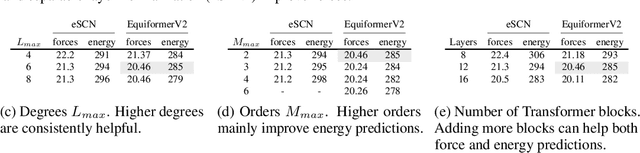
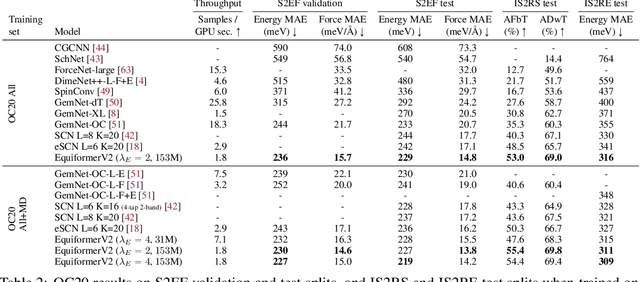
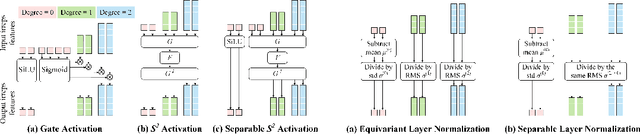
Equivariant Transformers such as Equiformer have demonstrated the efficacy of applying Transformers to the domain of 3D atomistic systems. However, they are still limited to small degrees of equivariant representations due to their computational complexity. In this paper, we investigate whether these architectures can scale well to higher degrees. Starting from Equiformer, we first replace $SO(3)$ convolutions with eSCN convolutions to efficiently incorporate higher-degree tensors. Then, to better leverage the power of higher degrees, we propose three architectural improvements -- attention re-normalization, separable $S^2$ activation and separable layer normalization. Putting this all together, we propose EquiformerV2, which outperforms previous state-of-the-art methods on the large-scale OC20 dataset by up to $12\%$ on forces, $4\%$ on energies, offers better speed-accuracy trade-offs, and $2\times$ reduction in DFT calculations needed for computing adsorption energies.
Spherical Channels for Modeling Atomic Interactions
Jun 29, 2022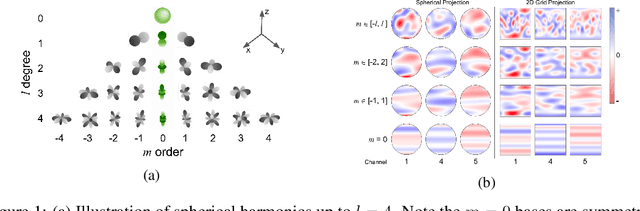
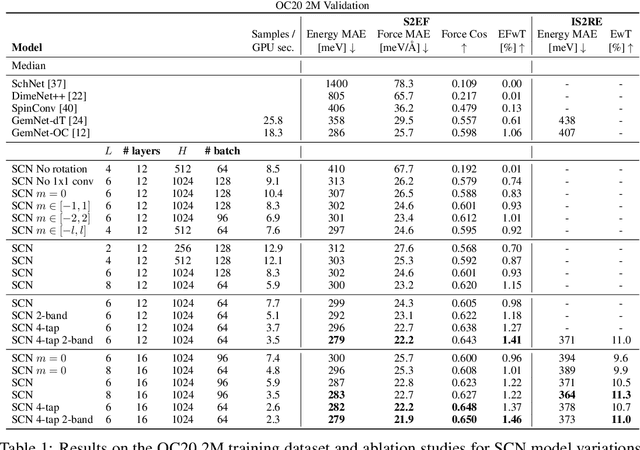
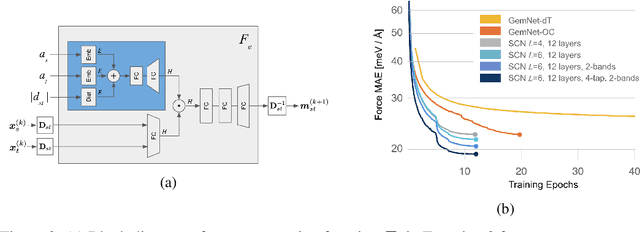
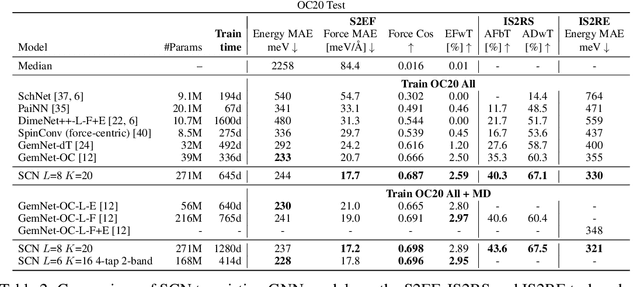
Modeling the energy and forces of atomic systems is a fundamental problem in computational chemistry with the potential to help address many of the world's most pressing problems, including those related to energy scarcity and climate change. These calculations are traditionally performed using Density Functional Theory, which is computationally very expensive. Machine learning has the potential to dramatically improve the efficiency of these calculations from days or hours to seconds. We propose the Spherical Channel Network (SCN) to model atomic energies and forces. The SCN is a graph neural network where nodes represent atoms and edges their neighboring atoms. The atom embeddings are a set of spherical functions, called spherical channels, represented using spherical harmonics. We demonstrate, that by rotating the embeddings based on the 3D edge orientation, more information may be utilized while maintaining the rotational equivariance of the messages. While equivariance is a desirable property, we find that by relaxing this constraint in both message passing and aggregation, improved accuracy may be achieved. We demonstrate state-of-the-art results on the large-scale Open Catalyst 2020 dataset in both energy and force prediction for numerous tasks and metrics.
The Open Catalyst 2020 Dataset and Community Challenges
Oct 20, 2020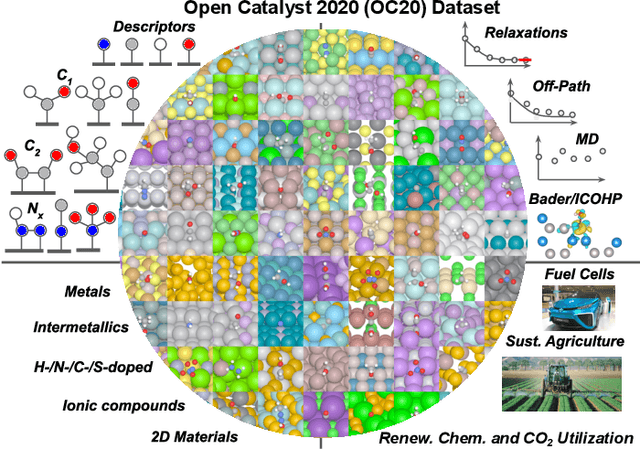

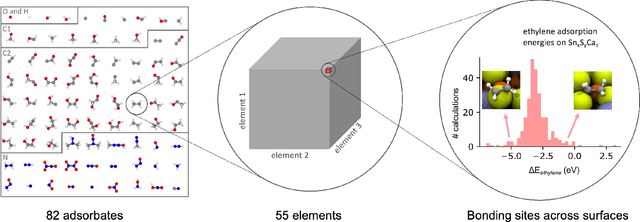
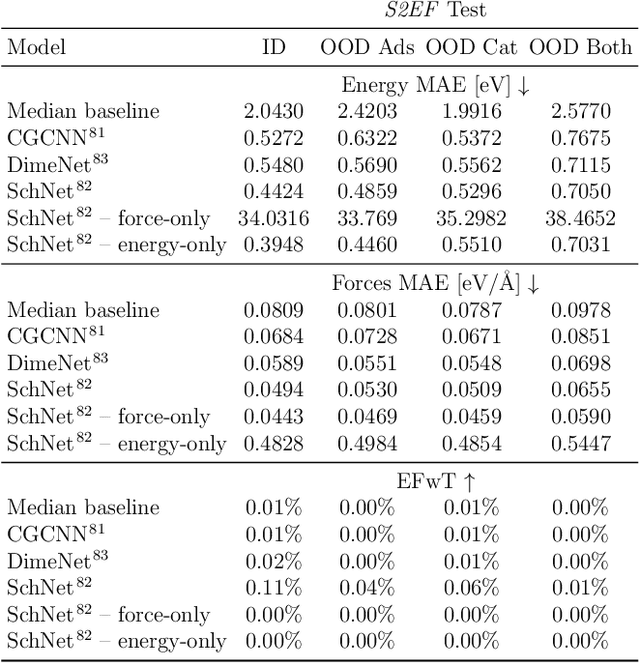
Catalyst discovery and optimization is key to solving many societal and energy challenges including solar fuels synthesis, long-term energy storage, and renewable fertilizer production. Despite considerable effort by the catalysis community to apply machine learning models to the computational catalyst discovery process, it remains an open challenge to build models that can generalize across both elemental compositions of surfaces and adsorbate identity/configurations, perhaps because datasets have been smaller in catalysis than related fields. To address this we developed the OC20 dataset, consisting of 1,281,121 Density Functional Theory (DFT) relaxations (264,900,500 single point evaluations) across a wide swath of materials, surfaces, and adsorbates (nitrogen, carbon, and oxygen chemistries). We supplemented this dataset with randomly perturbed structures, short timescale molecular dynamics, and electronic structure analyses. The dataset comprises three central tasks indicative of day-to-day catalyst modeling and comes with pre-defined train/validation/test splits to facilitate direct comparisons with future model development efforts. We applied three state-of-the-art graph neural network models (SchNet, Dimenet, CGCNN) to each of these tasks as baseline demonstrations for the community to build on. In almost every task, no upper limit on model size was identified, suggesting that even larger models are likely to improve on initial results. The dataset and baseline models are both provided as open resources, as well as a public leader board to encourage community contributions to solve these important tasks.
An Introduction to Electrocatalyst Design using Machine Learning for Renewable Energy Storage
Oct 14, 2020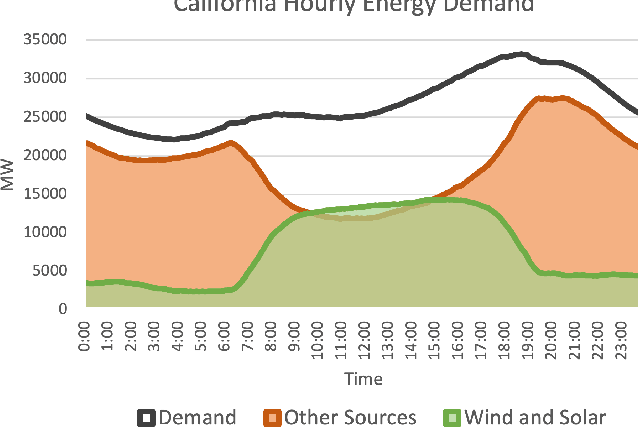
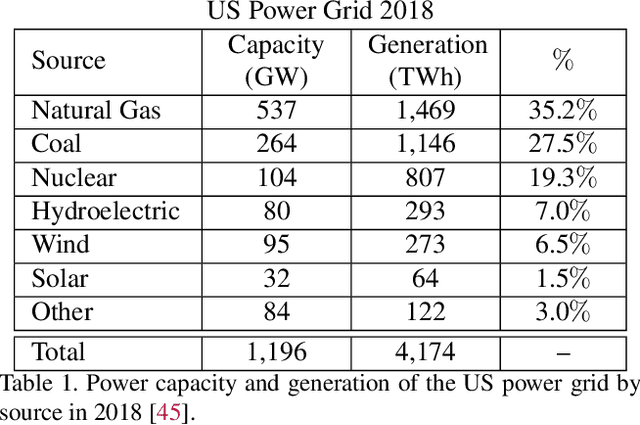
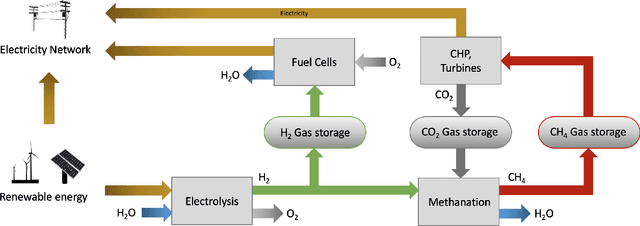
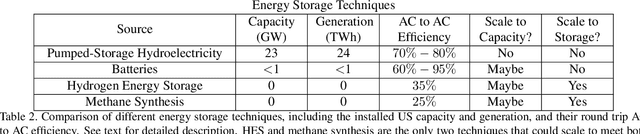
Scalable and cost-effective solutions to renewable energy storage are essential to addressing the world's rising energy needs while reducing climate change. As we increase our reliance on renewable energy sources such as wind and solar, which produce intermittent power, storage is needed to transfer power from times of peak generation to peak demand. This may require the storage of power for hours, days, or months. One solution that offers the potential of scaling to nation-sized grids is the conversion of renewable energy to other fuels, such as hydrogen or methane. To be widely adopted, this process requires cost-effective solutions to running electrochemical reactions. An open challenge is finding low-cost electrocatalysts to drive these reactions at high rates. Through the use of quantum mechanical simulations (density functional theory), new catalyst structures can be tested and evaluated. Unfortunately, the high computational cost of these simulations limits the number of structures that may be tested. The use of machine learning may provide a method to efficiently approximate these calculations, leading to new approaches in finding effective electrocatalysts. In this paper, we provide an introduction to the challenges in finding suitable electrocatalysts, how machine learning may be applied to the problem, and the use of the Open Catalyst Project OC20 dataset for model training.