 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe Open Catalyst 2022 Dataset and Challenges for Oxide Electrocatalysis
Paper and Code
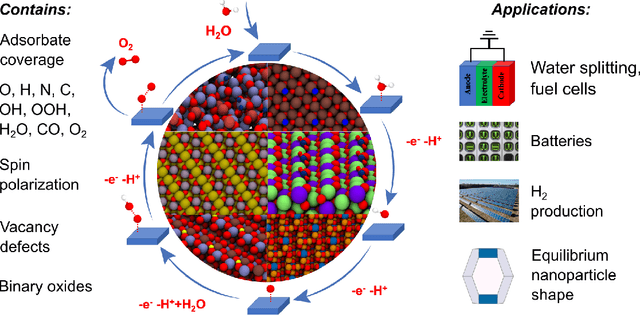
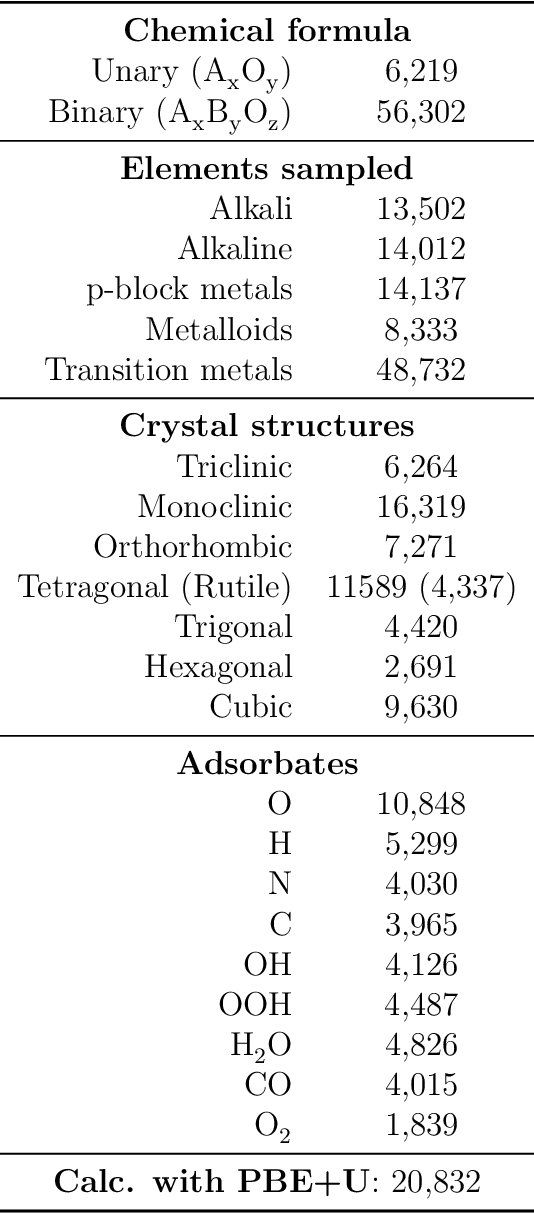
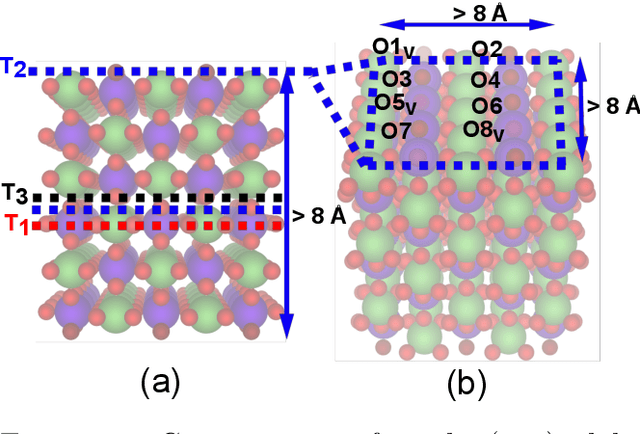
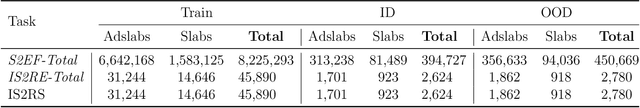
Computational catalysis and machine learning communities have made considerable progress in developing machine learning models for catalyst discovery and design. Yet, a general machine learning potential that spans the chemical space of catalysis is still out of reach. A significant hurdle is obtaining access to training data across a wide range of materials. One important class of materials where data is lacking are oxides, which inhibits models from studying the Oxygen Evolution Reaction and oxide electrocatalysis more generally. To address this we developed the Open Catalyst 2022(OC22) dataset, consisting of 62,521 Density Functional Theory (DFT) relaxations (~9,884,504 single point calculations) across a range of oxide materials, coverages, and adsorbates (*H, *O, *N, *C, *OOH, *OH, *OH2, *O2, *CO). We define generalized tasks to predict the total system energy that are applicable across catalysis, develop baseline performance of several graph neural networks (SchNet, DimeNet++, ForceNet, SpinConv, PaiNN, GemNet-dT, GemNet-OC), and provide pre-defined dataset splits to establish clear benchmarks for future efforts. For all tasks, we study whether combining datasets leads to better results, even if they contain different materials or adsorbates. Specifically, we jointly train models on Open Catalyst 2020 (OC20) Dataset and OC22, or fine-tune pretrained OC20 models on OC22. In the most general task, GemNet-OC sees a ~32% improvement in energy predictions through fine-tuning and a ~9% improvement in force predictions via joint training. Surprisingly, joint training on both the OC20 and much smaller OC22 datasets also improves total energy predictions on OC20 by ~19%. The dataset and baseline models are open sourced, and a public leaderboard will follow to encourage continued community developments on the total energy tasks and data.