 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe Open DAC 2023 Dataset and Challenges for Sorbent Discovery in Direct Air Capture
Nov 01, 2023
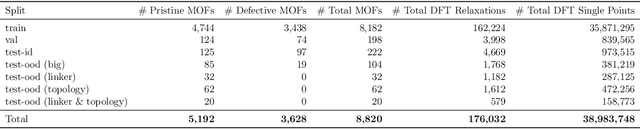
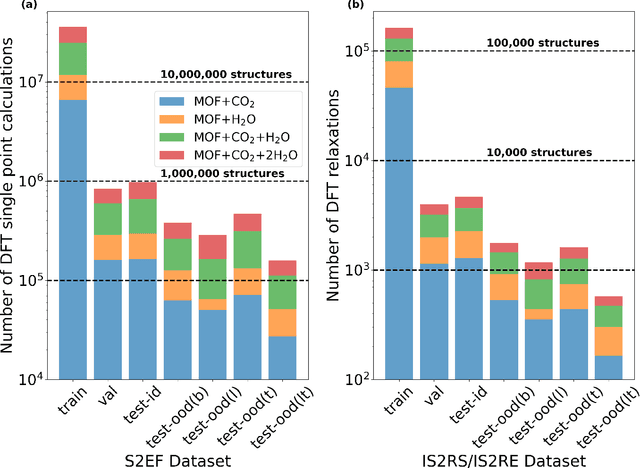

New methods for carbon dioxide removal are urgently needed to combat global climate change. Direct air capture (DAC) is an emerging technology to capture carbon dioxide directly from ambient air. Metal-organic frameworks (MOFs) have been widely studied as potentially customizable adsorbents for DAC. However, discovering promising MOF sorbents for DAC is challenging because of the vast chemical space to explore and the need to understand materials as functions of humidity and temperature. We explore a computational approach benefiting from recent innovations in machine learning (ML) and present a dataset named Open DAC 2023 (ODAC23) consisting of more than 38M density functional theory (DFT) calculations on more than 8,800 MOF materials containing adsorbed CO2 and/or H2O. ODAC23 is by far the largest dataset of MOF adsorption calculations at the DFT level of accuracy currently available. In addition to probing properties of adsorbed molecules, the dataset is a rich source of information on structural relaxation of MOFs, which will be useful in many contexts beyond specific applications for DAC. A large number of MOFs with promising properties for DAC are identified directly in ODAC23. We also trained state-of-the-art ML models on this dataset to approximate calculations at the DFT level. This open-source dataset and our initial ML models will provide an important baseline for future efforts to identify MOFs for a wide range of applications, including DAC.
Maximum-likelihood Estimators in Physics-Informed Neural Networks for High-dimensional Inverse Problems
Apr 14, 2023

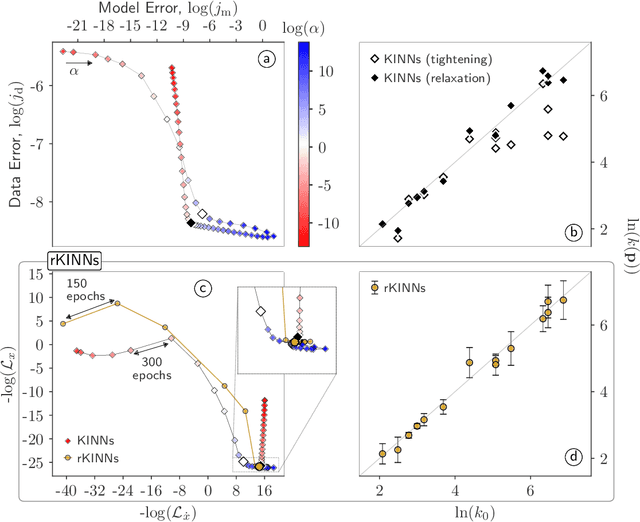

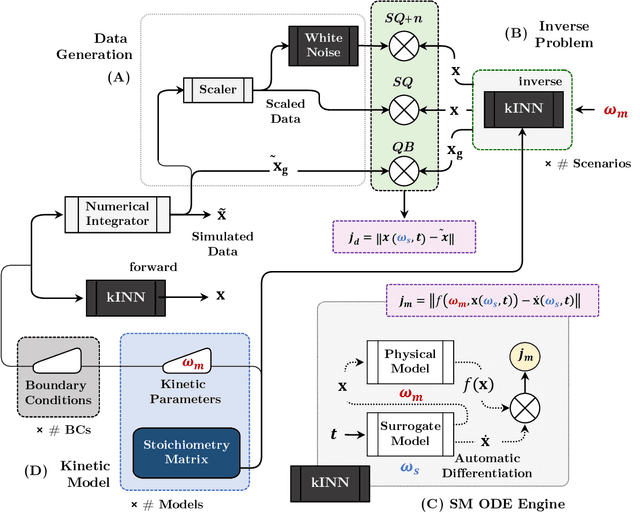
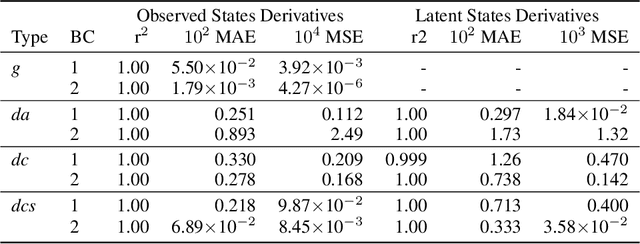
Physics-informed neural networks (PINNs) have proven a suitable mathematical scaffold for solving inverse ordinary (ODE) and partial differential equations (PDE). Typical inverse PINNs are formulated as soft-constrained multi-objective optimization problems with several hyperparameters. In this work, we demonstrate that inverse PINNs can be framed in terms of maximum-likelihood estimators (MLE) to allow explicit error propagation from interpolation to the physical model space through Taylor expansion, without the need of hyperparameter tuning. We explore its application to high-dimensional coupled ODEs constrained by differential algebraic equations that are common in transient chemical and biological kinetics. Furthermore, we show that singular-value decomposition (SVD) of the ODE coupling matrices (reaction stoichiometry matrix) provides reduced uncorrelated subspaces in which PINNs solutions can be represented and over which residuals can be projected. Finally, SVD bases serve as preconditioners for the inversion of covariance matrices in this hyperparameter-free robust application of MLE to ``kinetics-informed neural networks''.
A Priori Calibration of Transient Kinetics Data via Machine Learning
Sep 27, 2021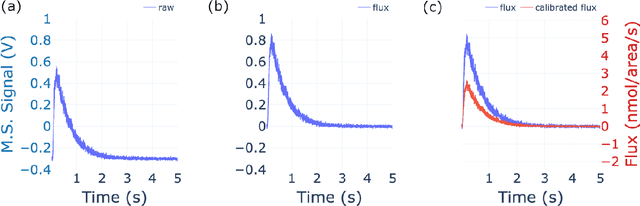
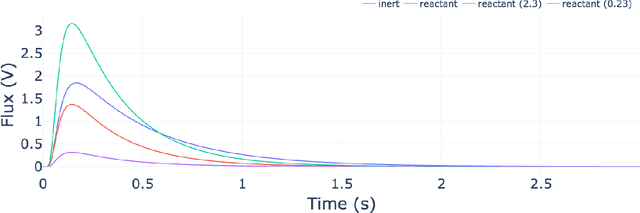
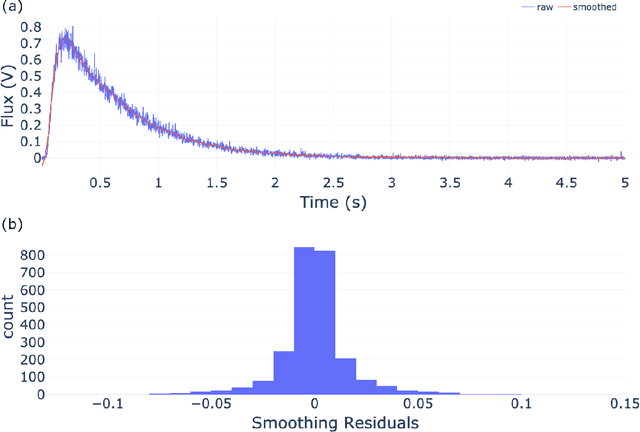
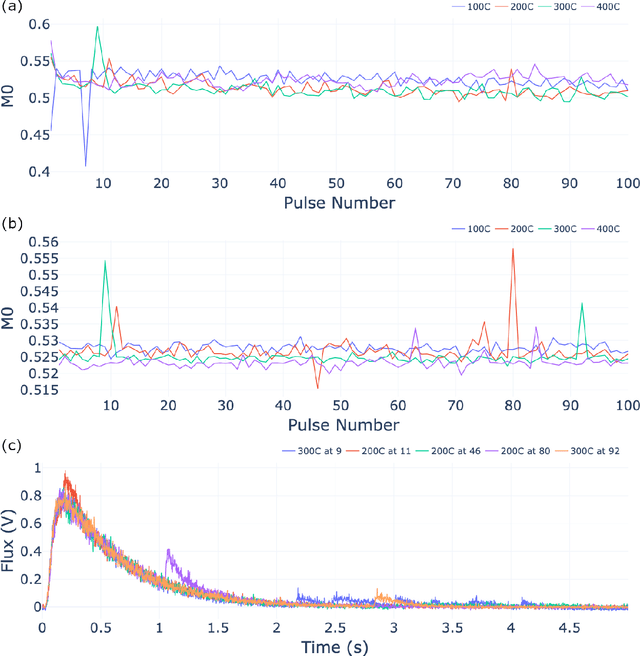
The temporal analysis of products reactor provides a vast amount of transient kinetic information that may be used to describe a variety of chemical features including the residence time distribution, kinetic coefficients, number of active sites, and the reaction mechanism. However, as with any measurement device, the TAP reactor signal is convoluted with noise. To reduce the uncertainty of the kinetic measurement and any derived parameters or mechanisms, proper preprocessing must be performed prior to any advanced analysis. This preprocessing consists of baseline correction, i.e., a shift in the voltage response, and calibration, i.e., a scaling of the flux response based on prior experiments. The current methodology of preprocessing requires significant user discretion and reliance on previous experiments that may drift over time. Herein we use machine learning techniques combined with physical constraints to convert the raw instrument signal to chemical information. As such, the proposed methodology demonstrates clear benefits over the traditional preprocessing in the calibration of the inert and feed mixture products without need of prior calibration experiments or heuristic input from the user.
A Universal Framework for Featurization of Atomistic Systems
Feb 05, 2021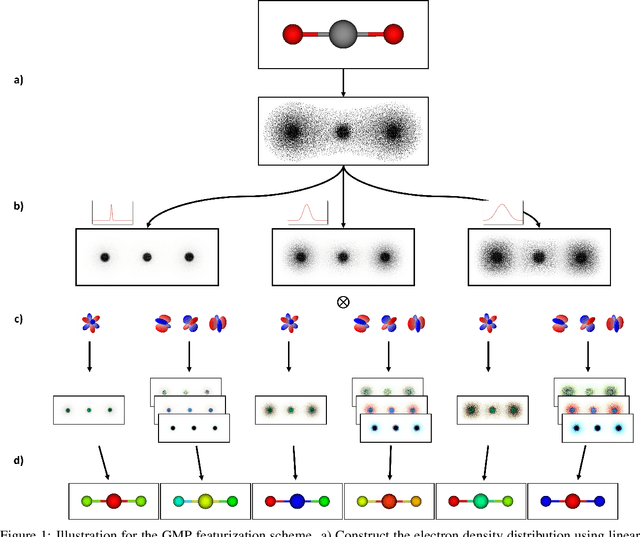

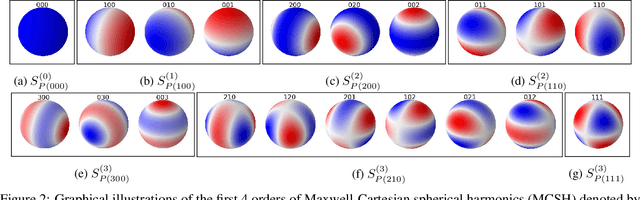
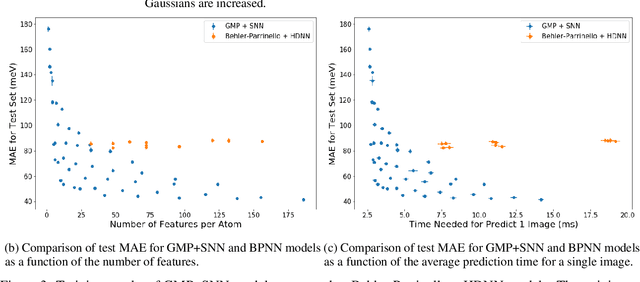
Molecular dynamics simulations are an invaluable tool in numerous scientific fields. However, the ubiquitous classical force fields cannot describe reactive systems, and quantum molecular dynamics are too computationally demanding to treat large systems or long timescales. Reactive force fields based on physics or machine learning can be used to bridge the gap in time and length scales, but these force fields require substantial effort to construct and are highly specific to given chemical composition and application. The extreme flexibility of machine learning models promises to yield reactive force fields that provide a more general description of chemical bonding. However, a significant limitation of machine learning models is the use of element-specific features, leading to models that scale poorly with the number of elements. This work introduces the Gaussian multi-pole (GMP) featurization scheme that utilizes physically-relevant multi-pole expansions of the electron density around atoms to yield feature vectors that interpolate between element types and have a fixed dimension regardless of the number of elements present. We combine GMP with neural networks to directly compare it to the widely-used Behler-Parinello symmetry functions for the MD17 dataset, revealing that it exhibits improved accuracy and computational efficiency. Further, we demonstrate that GMP-based models can achieve chemical accuracy for the QM9 dataset, and their accuracy remains reasonable even when extrapolating to new elements. Finally, we test GMP-based models for the Open Catalysis Project (OCP) dataset, revealing comparable performance and improved learning rates when compared to graph convolutional deep learning models. The results indicate that this featurization scheme fills a critical gap in the construction of efficient and transferable reactive force fields.
Kinetics-Informed Neural Networks
Nov 30, 2020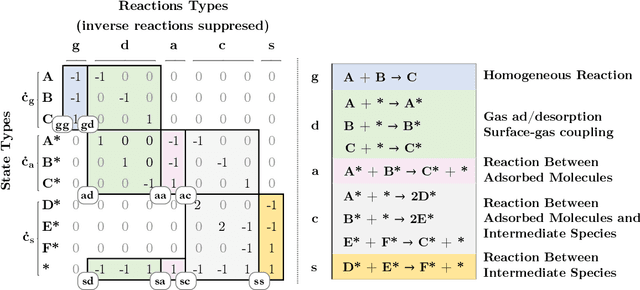
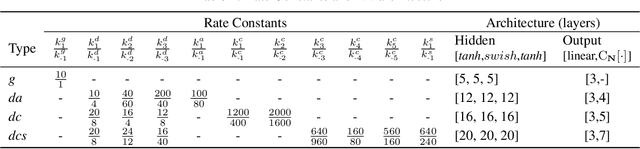
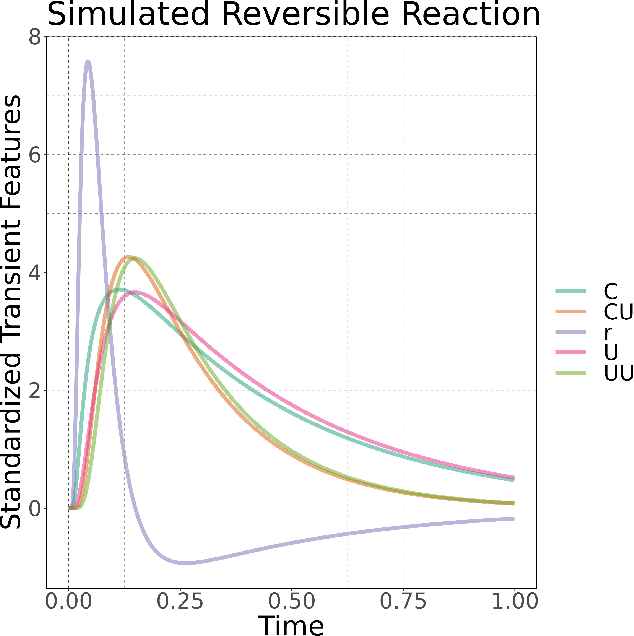
Chemical kinetics consists of the phenomenological framework for the disentanglement of reaction mechanisms, optimization of reaction performance and the rational design of chemical processes. Here, we utilize feed-forward artificial neural networks as basis functions for the construction of surrogate models to solve ordinary differential equations (ODEs) that describe microkinetic models (MKMs). We present an algebraic framework for the mathematical description and classification of reaction networks, types of elementary reaction, and chemical species. Under this framework, we demonstrate that the simultaneous training of neural nets and kinetic model parameters in a regularized multiobjective optimization setting leads to the solution of the inverse problem through the estimation of kinetic parameters from synthetic experimental data. We probe the limits at which kinetic parameters can be retrieved as a function of knowledge about the chemical system states over time, and assess the robustness of the methodology with respect to statistical noise. This surrogate approach to inverse kinetic ODEs can assist in the elucidation of reaction mechanisms based on transient data.
Data Driven Reaction Mechanism Estimation via Transient Kinetics and Machine Learning
Nov 17, 2020

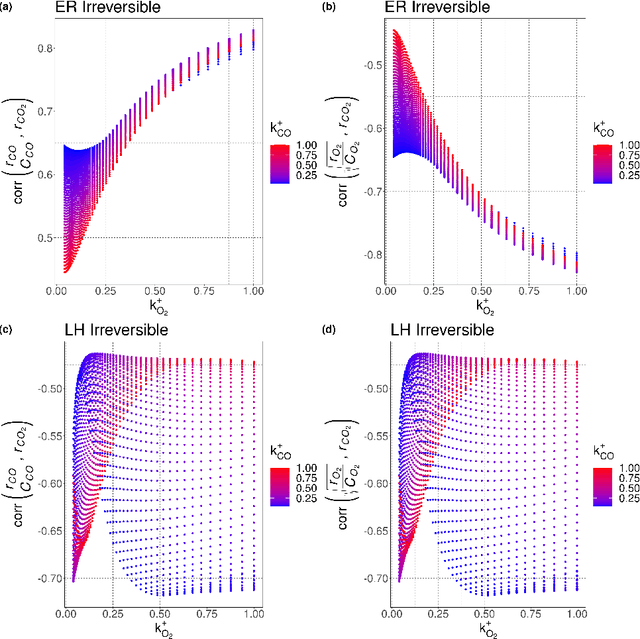
Understanding the set of elementary steps and kinetics in each reaction is extremely valuable to make informed decisions about creating the next generation of catalytic materials. With physical and mechanistic complexity of industrial catalysts, it is critical to obtain kinetic information through experimental methods. As such, this work details a methodology based on the combination of transient rate/concentration dependencies and machine learning to measure the number of active sites, the individual rate constants, and gain insight into the mechanism under a complex set of elementary steps. This new methodology was applied to simulated transient responses to verify its ability to obtain correct estimates of the micro-kinetic coefficients. Furthermore, experimental CO oxidation data was analyzed to reveal the Langmuir-Hinshelwood mechanism driving the reaction. As oxygen accumulated on the catalyst, a transition in the mechanism was clearly defined in the machine learning analysis due to the large amount of kinetic information available from transient reaction techniques. This methodology is proposed as a new data driven approach to characterize how materials control complex reaction mechanisms relying exclusively on experimental data.
ElectroLens: Understanding Atomistic Simulations Through Spatially-resolved Visualization of High-dimensional Features
Aug 20, 2019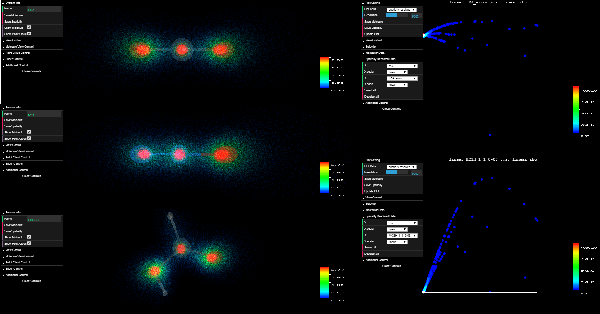
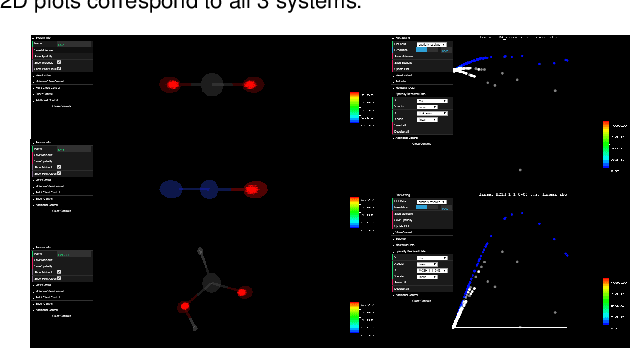
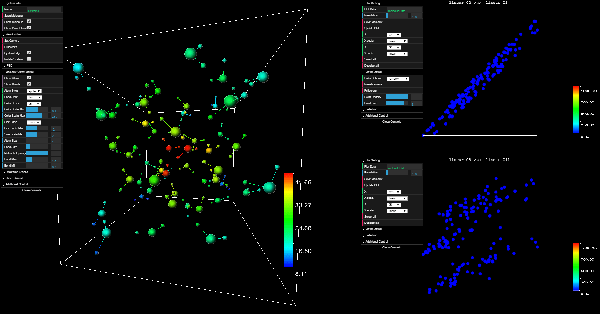
In recent years, machine learning (ML) has gained significant popularity in the field of chemical informatics and electronic structure theory. These techniques often require researchers to engineer abstract "features" that encode chemical concepts into a mathematical form compatible with the input to machine-learning models. However, there is no existing tool to connect these abstract features back to the actual chemical system, making it difficult to diagnose failures and to build intuition about the meaning of the features. We present ElectroLens, a new visualization tool for high-dimensional spatially-resolved features to tackle this problem. The tool visualizes high-dimensional data sets for atomistic and electron environment features by a series of linked 3D views and 2D plots. The tool is able to connect different derived features and their corresponding regions in 3D via interactive selection. It is built to be scalable, and integrate with existing infrastructure.