 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe Role of Reference Points in Machine-Learned Atomistic Simulation Models
Oct 28, 2023



This paper introduces the Chemical Environment Modeling Theory (CEMT), a novel, generalized framework designed to overcome the limitations inherent in traditional atom-centered Machine Learning Force Field (MLFF) models, widely used in atomistic simulations of chemical systems. CEMT demonstrated enhanced flexibility and adaptability by allowing reference points to exist anywhere within the modeled domain and thus, enabling the study of various model architectures. Utilizing Gaussian Multipole (GMP) featurization functions, several models with different reference point sets, including finite difference grid-centered and bond-centered models, were tested to analyze the variance in capabilities intrinsic to models built on distinct reference points. The results underscore the potential of non-atom-centered reference points in force training, revealing variations in prediction accuracy, inference speed and learning efficiency. Finally, a unique connection between CEMT and real-space orbital-free finite element Density Functional Theory (FE-DFT) is established, and the implications include the enhancement of data efficiency and robustness. It allows the leveraging of spatially-resolved energy densities and charge densities from FE-DFT calculations, as well as serving as a pivotal step towards integrating known quantum-mechanical laws into the architecture of ML models.
Lessons in Reproducibility: Insights from NLP Studies in Materials Science
Jul 28, 2023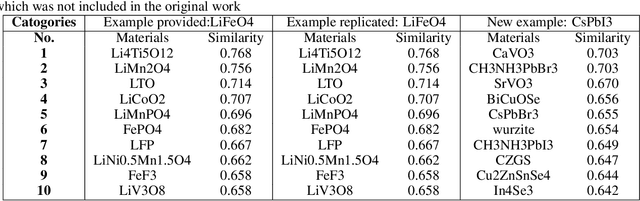
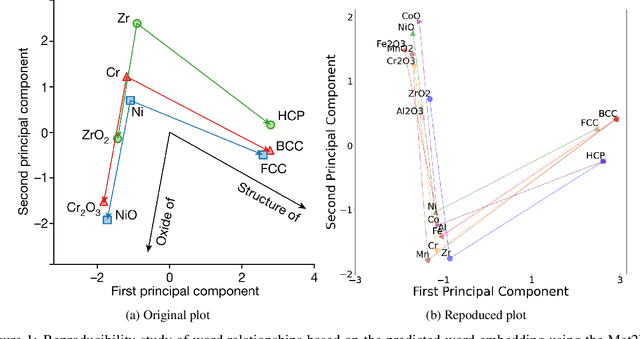
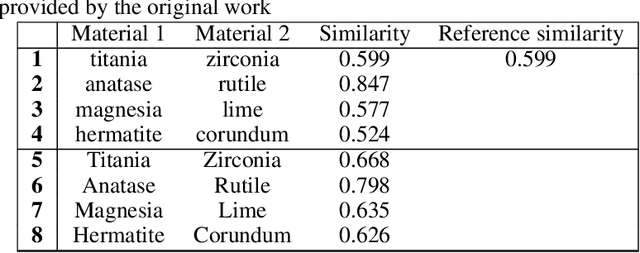

Natural Language Processing (NLP), a cornerstone field within artificial intelligence, has been increasingly utilized in the field of materials science literature. Our study conducts a reproducibility analysis of two pioneering works within this domain: "Machine-learned and codified synthesis parameters of oxide materials" by Kim et al., and "Unsupervised word embeddings capture latent knowledge from materials science literature" by Tshitoyan et al. We aim to comprehend these studies from a reproducibility perspective, acknowledging their significant influence on the field of materials informatics, rather than critiquing them. Our study indicates that both papers offered thorough workflows, tidy and well-documented codebases, and clear guidance for model evaluation. This makes it easier to replicate their results successfully and partially reproduce their findings. In doing so, they set commendable standards for future materials science publications to aspire to. However, our analysis also highlights areas for improvement such as to provide access to training data where copyright restrictions permit, more transparency on model architecture and the training process, and specifications of software dependency versions. We also cross-compare the word embedding models between papers, and find that some key differences in reproducibility and cross-compatibility are attributable to design choices outside the bounds of the models themselves. In summary, our study appreciates the benchmark set by these seminal papers while advocating for further enhancements in research reproducibility practices in the field of NLP for materials science. This balance of understanding and continuous improvement will ultimately propel the intersecting domains of NLP and materials science literature into a future of exciting discoveries.
A Universal Framework for Featurization of Atomistic Systems
Feb 05, 2021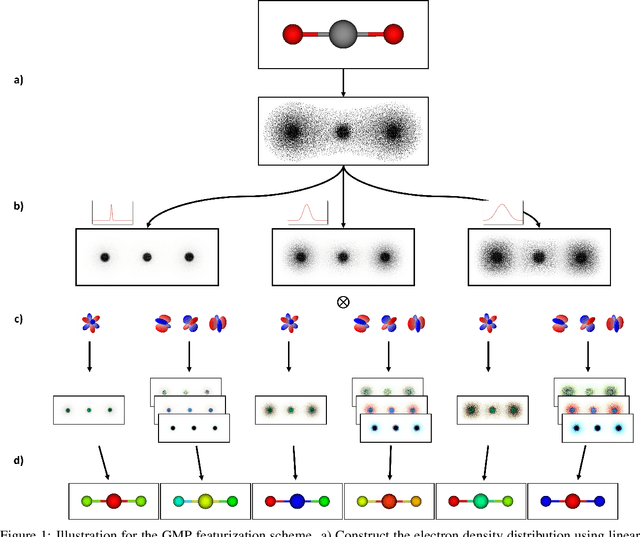

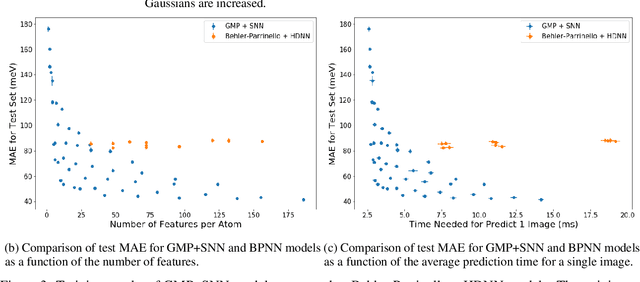
Molecular dynamics simulations are an invaluable tool in numerous scientific fields. However, the ubiquitous classical force fields cannot describe reactive systems, and quantum molecular dynamics are too computationally demanding to treat large systems or long timescales. Reactive force fields based on physics or machine learning can be used to bridge the gap in time and length scales, but these force fields require substantial effort to construct and are highly specific to given chemical composition and application. The extreme flexibility of machine learning models promises to yield reactive force fields that provide a more general description of chemical bonding. However, a significant limitation of machine learning models is the use of element-specific features, leading to models that scale poorly with the number of elements. This work introduces the Gaussian multi-pole (GMP) featurization scheme that utilizes physically-relevant multi-pole expansions of the electron density around atoms to yield feature vectors that interpolate between element types and have a fixed dimension regardless of the number of elements present. We combine GMP with neural networks to directly compare it to the widely-used Behler-Parinello symmetry functions for the MD17 dataset, revealing that it exhibits improved accuracy and computational efficiency. Further, we demonstrate that GMP-based models can achieve chemical accuracy for the QM9 dataset, and their accuracy remains reasonable even when extrapolating to new elements. Finally, we test GMP-based models for the Open Catalysis Project (OCP) dataset, revealing comparable performance and improved learning rates when compared to graph convolutional deep learning models. The results indicate that this featurization scheme fills a critical gap in the construction of efficient and transferable reactive force fields.
ElectroLens: Understanding Atomistic Simulations Through Spatially-resolved Visualization of High-dimensional Features
Aug 20, 2019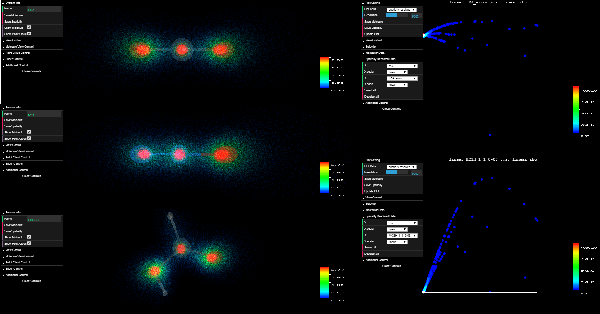
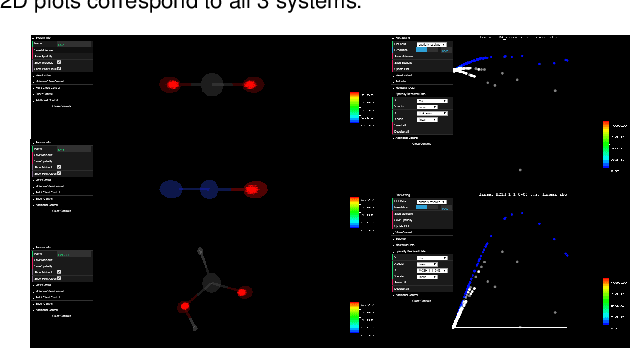
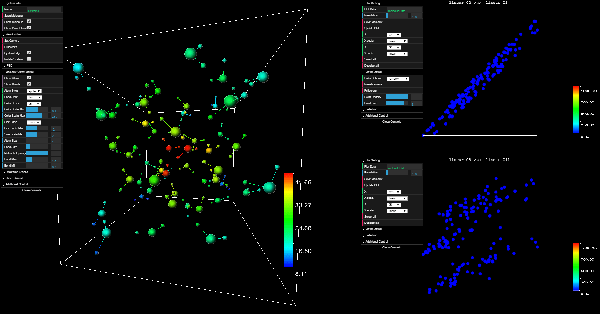
In recent years, machine learning (ML) has gained significant popularity in the field of chemical informatics and electronic structure theory. These techniques often require researchers to engineer abstract "features" that encode chemical concepts into a mathematical form compatible with the input to machine-learning models. However, there is no existing tool to connect these abstract features back to the actual chemical system, making it difficult to diagnose failures and to build intuition about the meaning of the features. We present ElectroLens, a new visualization tool for high-dimensional spatially-resolved features to tackle this problem. The tool visualizes high-dimensional data sets for atomistic and electron environment features by a series of linked 3D views and 2D plots. The tool is able to connect different derived features and their corresponding regions in 3D via interactive selection. It is built to be scalable, and integrate with existing infrastructure.