 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFINETUNA: Fine-tuning Accelerated Molecular Simulations
Paper and Code
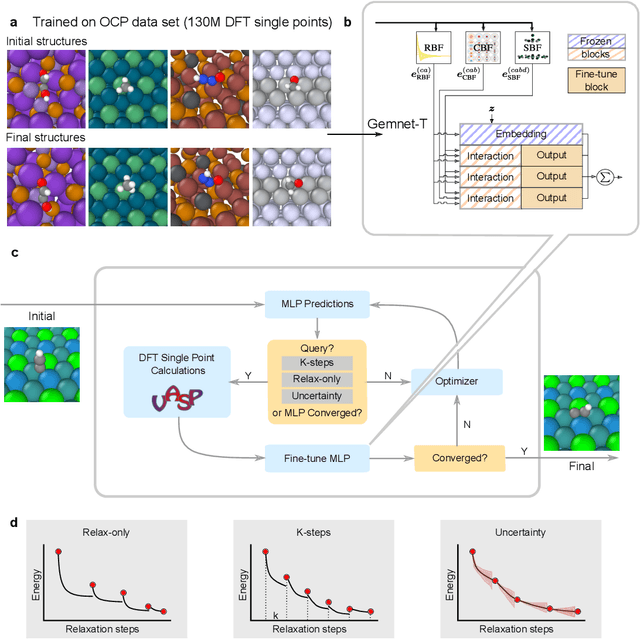
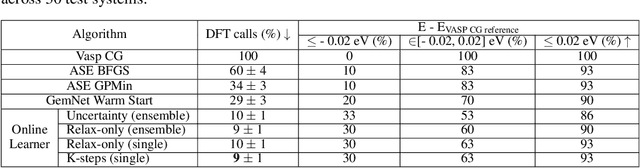
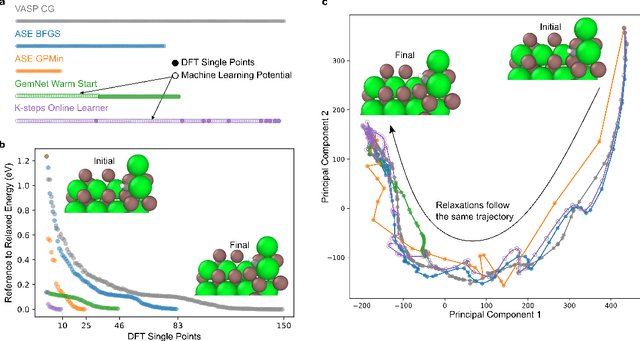
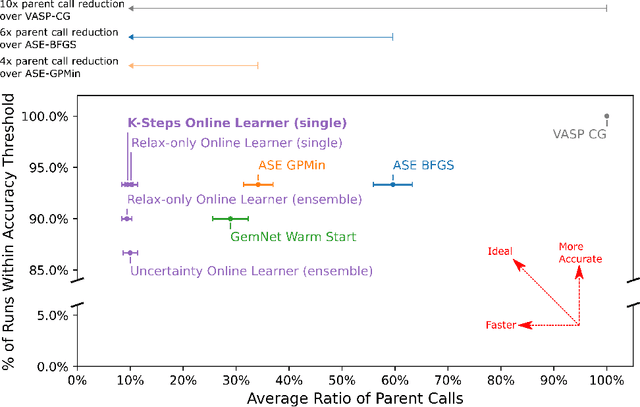
Machine learning approaches have the potential to approximate Density Functional Theory (DFT) for atomistic simulations in a computationally efficient manner, which could dramatically increase the impact of computational simulations on real-world problems. However, they are limited by their accuracy and the cost of generating labeled data. Here, we present an online active learning framework for accelerating the simulation of atomic systems efficiently and accurately by incorporating prior physical information learned by large-scale pre-trained graph neural network models from the Open Catalyst Project. Accelerating these simulations enables useful data to be generated more cheaply, allowing better models to be trained and more atomistic systems to be screened. We also present a method of comparing local optimization techniques on the basis of both their speed and accuracy. Experiments on 30 benchmark adsorbate-catalyst systems show that our method of transfer learning to incorporate prior information from pre-trained models accelerates simulations by reducing the number of DFT calculations by 91%, while meeting an accuracy threshold of 0.02 eV 93% of the time. Finally, we demonstrate a technique for leveraging the interactive functionality built in to VASP to efficiently compute single point calculations within our online active learning framework without the significant startup costs. This allows VASP to work in tandem with our framework while requiring 75% fewer self-consistent cycles than conventional single point calculations. The online active learning implementation, and examples using the VASP interactive code, are available in the open source FINETUNA package on Github.