 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeQT-Net: Rethinking Evaluation of AI Models in Atomic Chemical Space
May 11, 2026Atomic properties such as partial charges or multipoles encode chemically meaningful information that can inform downstream molecular property prediction, but their evaluation as machine learning targets has been complicated by the absence of a principled out-of-distribution evaluation protocol at the atomic level. In this work, we propose a held-out evaluation protocol that clusters atomic environments by SOAP descriptors and computes metrics accounting only for cluster labels unseen during training. Following this procedure, we use 5$\times$5 cross-validation and Tukey's HSD to run a statistically rigorous comparison of E(3)-equivariant against non-equivariant, rotationally augmented models for predicting electron populations and multipoles of H, C, N, and O atoms. Building on our results, we introduce the Quantum Topological Neural Network (QT-Net), a rotationally augmented, non-equivariant graph neural network. We show that QT-Net can be used to infer properties of atoms in molecules from QM9 outside our training set, and that these inferred properties can yield improvement when used as input features for downstream molecular property prediction. To further validate the framework, molecular dipole moments computed from QT-Net's per-atom outputs recover the ground-truth values reported in QM9. We release all code and data, including a JAX implementation of QT-Net, to support the broader use of learned QTA properties as inductive biases for atomic-scale molecular machine learning.
SymmCD: Symmetry-Preserving Crystal Generation with Diffusion Models
Feb 05, 2025Generating novel crystalline materials has potential to lead to advancements in fields such as electronics, energy storage, and catalysis. The defining characteristic of crystals is their symmetry, which plays a central role in determining their physical properties. However, existing crystal generation methods either fail to generate materials that display the symmetries of real-world crystals, or simply replicate the symmetry information from examples in a database. To address this limitation, we propose SymmCD, a novel diffusion-based generative model that explicitly incorporates crystallographic symmetry into the generative process. We decompose crystals into two components and learn their joint distribution through diffusion: 1) the asymmetric unit, the smallest subset of the crystal which can generate the whole crystal through symmetry transformations, and; 2) the symmetry transformations needed to be applied to each atom in the asymmetric unit. We also use a novel and interpretable representation for these transformations, enabling generalization across different crystallographic symmetry groups. We showcase the competitive performance of SymmCD on a subset of the Materials Project, obtaining diverse and valid crystals with realistic symmetries and predicted properties.
Energy & Force Regression on DFT Trajectories is Not Enough for Universal Machine Learning Interatomic Potentials
Feb 05, 2025



Universal Machine Learning Interactomic Potentials (MLIPs) enable accelerated simulations for materials discovery. However, current research efforts fail to impactfully utilize MLIPs due to: 1. Overreliance on Density Functional Theory (DFT) for MLIP training data creation; 2. MLIPs' inability to reliably and accurately perform large-scale molecular dynamics (MD) simulations for diverse materials; 3. Limited understanding of MLIPs' underlying capabilities. To address these shortcomings, we aargue that MLIP research efforts should prioritize: 1. Employing more accurate simulation methods for large-scale MLIP training data creation (e.g. Coupled Cluster Theory) that cover a wide range of materials design spaces; 2. Creating MLIP metrology tools that leverage large-scale benchmarking, visualization, and interpretability analyses to provide a deeper understanding of MLIPs' inner workings; 3. Developing computationally efficient MLIPs to execute MD simulations that accurately model a broad set of materials properties. Together, these interdisciplinary research directions can help further the real-world application of MLIPs to accurately model complex materials at device scale.
Stiefel Flow Matching for Moment-Constrained Structure Elucidation
Dec 17, 2024



Molecular structure elucidation is a fundamental step in understanding chemical phenomena, with applications in identifying molecules in natural products, lab syntheses, forensic samples, and the interstellar medium. We consider the task of predicting a molecule's all-atom 3D structure given only its molecular formula and moments of inertia, motivated by the ability of rotational spectroscopy to measure these moments. While existing generative models can conditionally sample 3D structures with approximately correct moments, this soft conditioning fails to leverage the many digits of precision afforded by experimental rotational spectroscopy. To address this, we first show that the space of $n$-atom point clouds with a fixed set of moments of inertia is embedded in the Stiefel manifold $\mathrm{St}(n, 4)$. We then propose Stiefel Flow Matching as a generative model for elucidating 3D structure under exact moment constraints. Additionally, we learn simpler and shorter flows by finding approximate solutions for equivariant optimal transport on the Stiefel manifold. Empirically, enforcing exact moment constraints allows Stiefel Flow Matching to achieve higher success rates and faster sampling than Euclidean diffusion models, even on high-dimensional manifolds corresponding to large molecules in the GEOM dataset.
Foundational Large Language Models for Materials Research
Dec 12, 2024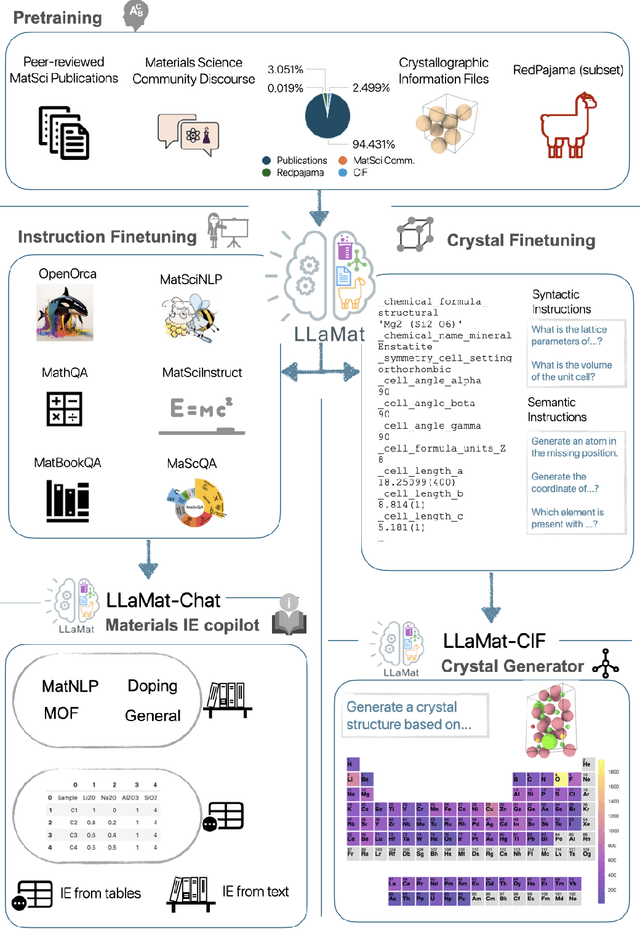
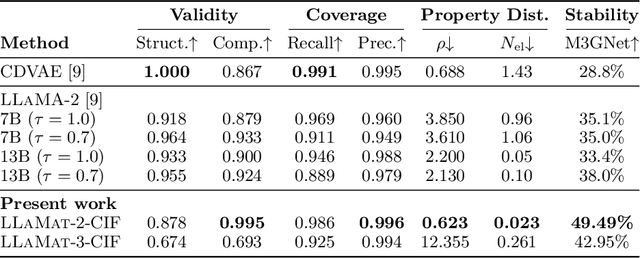
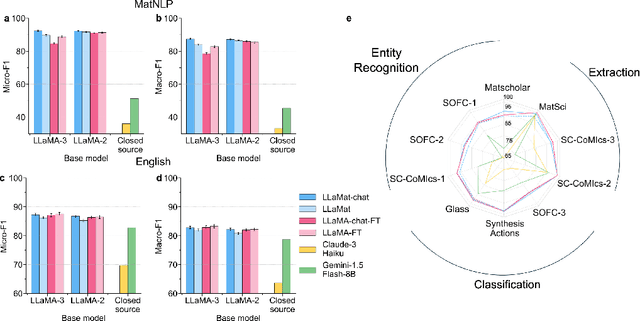
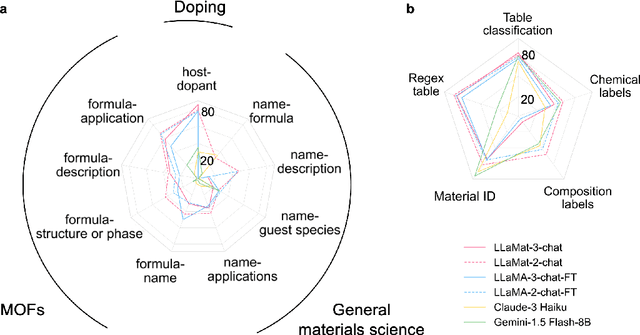
Materials discovery and development are critical for addressing global challenges. Yet, the exponential growth in materials science literature comprising vast amounts of textual data has created significant bottlenecks in knowledge extraction, synthesis, and scientific reasoning. Large Language Models (LLMs) offer unprecedented opportunities to accelerate materials research through automated analysis and prediction. Still, their effective deployment requires domain-specific adaptation for understanding and solving domain-relevant tasks. Here, we present LLaMat, a family of foundational models for materials science developed through continued pretraining of LLaMA models on an extensive corpus of materials literature and crystallographic data. Through systematic evaluation, we demonstrate that LLaMat excels in materials-specific NLP and structured information extraction while maintaining general linguistic capabilities. The specialized LLaMat-CIF variant demonstrates unprecedented capabilities in crystal structure generation, predicting stable crystals with high coverage across the periodic table. Intriguingly, despite LLaMA-3's superior performance in comparison to LLaMA-2, we observe that LLaMat-2 demonstrates unexpectedly enhanced domain-specific performance across diverse materials science tasks, including structured information extraction from text and tables, more particularly in crystal structure generation, a potential adaptation rigidity in overtrained LLMs. Altogether, the present work demonstrates the effectiveness of domain adaptation towards developing practically deployable LLM copilots for materials research. Beyond materials science, our findings reveal important considerations for domain adaptation of LLMs, such as model selection, training methodology, and domain-specific performance, which may influence the development of specialized scientific AI systems.
MatExpert: Decomposing Materials Discovery by Mimicking Human Experts
Oct 26, 2024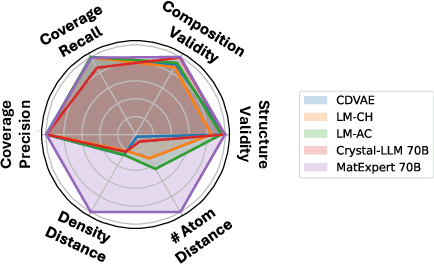
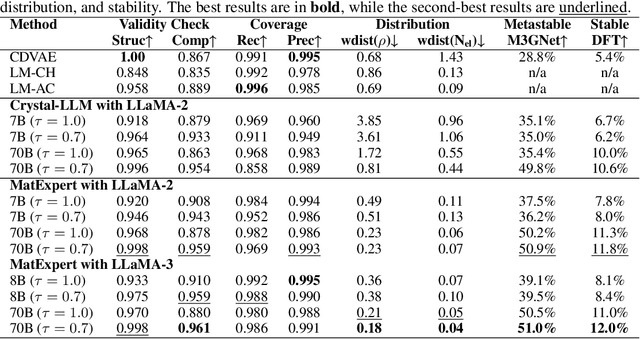

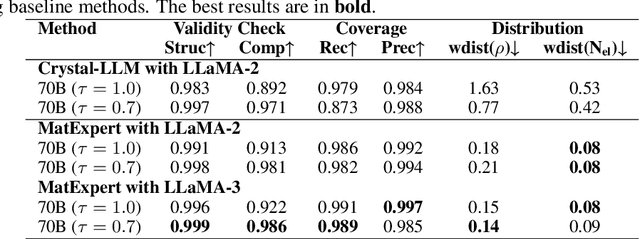
Material discovery is a critical research area with profound implications for various industries. In this work, we introduce MatExpert, a novel framework that leverages Large Language Models (LLMs) and contrastive learning to accelerate the discovery and design of new solid-state materials. Inspired by the workflow of human materials design experts, our approach integrates three key stages: retrieval, transition, and generation. First, in the retrieval stage, MatExpert identifies an existing material that closely matches the desired criteria. Second, in the transition stage, MatExpert outlines the necessary modifications to transform this material formulation to meet specific requirements outlined by the initial user query. Third, in the generation state, MatExpert performs detailed computations and structural generation to create new materials based on the provided information. Our experimental results demonstrate that MatExpert outperforms state-of-the-art methods in material generation tasks, achieving superior performance across various metrics including validity, distribution, and stability. As such, MatExpert represents a meaningful advancement in computational material discovery using langauge-based generative models.
Deconstructing equivariant representations in molecular systems
Oct 10, 2024Recent equivariant models have shown significant progress in not just chemical property prediction, but as surrogates for dynamical simulations of molecules and materials. Many of the top performing models in this category are built within the framework of tensor products, which preserves equivariance by restricting interactions and transformations to those that are allowed by symmetry selection rules. Despite being a core part of the modeling process, there has not yet been much attention into understanding what information persists in these equivariant representations, and their general behavior outside of benchmark metrics. In this work, we report on a set of experiments using a simple equivariant graph convolution model on the QM9 dataset, focusing on correlating quantitative performance with the resulting molecular graph embeddings. Our key finding is that, for a scalar prediction task, many of the irreducible representations are simply ignored during training -- specifically those pertaining to vector ($l=1$) and tensor quantities ($l=2$) -- an issue that does not necessarily make itself evident in the test metric. We empirically show that removing some unused orders of spherical harmonics improves model performance, correlating with improved latent space structure. We provide a number of recommendations for future experiments to try and improve efficiency and utilization of equivariant features based on these observations.
HoneyComb: A Flexible LLM-Based Agent System for Materials Science
Aug 29, 2024



The emergence of specialized large language models (LLMs) has shown promise in addressing complex tasks for materials science. Many LLMs, however, often struggle with distinct complexities of material science tasks, such as materials science computational tasks, and often rely heavily on outdated implicit knowledge, leading to inaccuracies and hallucinations. To address these challenges, we introduce HoneyComb, the first LLM-based agent system specifically designed for materials science. HoneyComb leverages a novel, high-quality materials science knowledge base (MatSciKB) and a sophisticated tool hub (ToolHub) to enhance its reasoning and computational capabilities tailored to materials science. MatSciKB is a curated, structured knowledge collection based on reliable literature, while ToolHub employs an Inductive Tool Construction method to generate, decompose, and refine API tools for materials science. Additionally, HoneyComb leverages a retriever module that adaptively selects the appropriate knowledge source or tools for specific tasks, thereby ensuring accuracy and relevance. Our results demonstrate that HoneyComb significantly outperforms baseline models across various tasks in materials science, effectively bridging the gap between current LLM capabilities and the specialized needs of this domain. Furthermore, our adaptable framework can be easily extended to other scientific domains, highlighting its potential for broad applicability in advancing scientific research and applications.
From Text to Insight: Large Language Models for Materials Science Data Extraction
Jul 23, 2024



The vast majority of materials science knowledge exists in unstructured natural language, yet structured data is crucial for innovative and systematic materials design. Traditionally, the field has relied on manual curation and partial automation for data extraction for specific use cases. The advent of large language models (LLMs) represents a significant shift, potentially enabling efficient extraction of structured, actionable data from unstructured text by non-experts. While applying LLMs to materials science data extraction presents unique challenges, domain knowledge offers opportunities to guide and validate LLM outputs. This review provides a comprehensive overview of LLM-based structured data extraction in materials science, synthesizing current knowledge and outlining future directions. We address the lack of standardized guidelines and present frameworks for leveraging the synergy between LLMs and materials science expertise. This work serves as a foundational resource for researchers aiming to harness LLMs for data-driven materials research. The insights presented here could significantly enhance how researchers across disciplines access and utilize scientific information, potentially accelerating the development of novel materials for critical societal needs.
MatText: Do Language Models Need More than Text & Scale for Materials Modeling?
Jun 25, 2024



Effectively representing materials as text has the potential to leverage the vast advancements of large language models (LLMs) for discovering new materials. While LLMs have shown remarkable success in various domains, their application to materials science remains underexplored. A fundamental challenge is the lack of understanding of how to best utilize text-based representations for materials modeling. This challenge is further compounded by the absence of a comprehensive benchmark to rigorously evaluate the capabilities and limitations of these text representations in capturing the complexity of material systems. To address this gap, we propose MatText, a suite of benchmarking tools and datasets designed to systematically evaluate the performance of language models in modeling materials. MatText encompasses nine distinct text-based representations for material systems, including several novel representations. Each representation incorporates unique inductive biases that capture relevant information and integrate prior physical knowledge about materials. Additionally, MatText provides essential tools for training and benchmarking the performance of language models in the context of materials science. These tools include standardized dataset splits for each representation, probes for evaluating sensitivity to geometric factors, and tools for seamlessly converting crystal structures into text. Using MatText, we conduct an extensive analysis of the capabilities of language models in modeling materials. Our findings reveal that current language models consistently struggle to capture the geometric information crucial for materials modeling across all representations. Instead, these models tend to leverage local information, which is emphasized in some of our novel representations. Our analysis underscores MatText's ability to reveal shortcomings of text-based methods for materials design.