 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSustainable Materials Discovery in the Era of Artificial Intelligence
Jan 29, 2026Artificial intelligence (AI) has transformed materials discovery, enabling rapid exploration of chemical space through generative models and surrogate screening. Yet current AI workflows optimize performance first, deferring sustainability to post synthesis assessment. This creates inefficiency by the time environmental burdens are quantified, resources have been invested in potentially unsustainable solutions. The disconnect between atomic scale design and lifecycle assessment (LCA) reflects fundamental challenges, data scarcity across heterogeneous sources, scale gaps from atoms to industrial systems, uncertainty in synthesis pathways, and the absence of frameworks that co-optimize performance with environmental impact. We propose to integrate upstream machine learning (ML) assisted materials discovery with downstream lifecycle assessment into a uniform ML-LCA environment. The framework ML-LCA integrates five components, information extraction for building materials-environment knowledge bases, harmonized databases linking properties to sustainability metrics, multi-scale models bridging atomic properties to lifecycle impacts, ensemble prediction of manufacturing pathways with uncertainty quantification, and uncertainty-aware optimization enabling simultaneous performance-sustainability navigation. Case studies spanning glass, cement, semiconductor photoresists, and polymers demonstrate both necessity and feasibility while identifying material-specific integration challenges. Realizing ML-LCA demands coordinated advances in data infrastructure, ex-ante assessment methodologies, multi-objective optimization, and regulatory alignment enabling the discovery of materials that are sustainable by design rather than by chance.
Energy & Force Regression on DFT Trajectories is Not Enough for Universal Machine Learning Interatomic Potentials
Feb 05, 2025



Universal Machine Learning Interactomic Potentials (MLIPs) enable accelerated simulations for materials discovery. However, current research efforts fail to impactfully utilize MLIPs due to: 1. Overreliance on Density Functional Theory (DFT) for MLIP training data creation; 2. MLIPs' inability to reliably and accurately perform large-scale molecular dynamics (MD) simulations for diverse materials; 3. Limited understanding of MLIPs' underlying capabilities. To address these shortcomings, we aargue that MLIP research efforts should prioritize: 1. Employing more accurate simulation methods for large-scale MLIP training data creation (e.g. Coupled Cluster Theory) that cover a wide range of materials design spaces; 2. Creating MLIP metrology tools that leverage large-scale benchmarking, visualization, and interpretability analyses to provide a deeper understanding of MLIPs' inner workings; 3. Developing computationally efficient MLIPs to execute MD simulations that accurately model a broad set of materials properties. Together, these interdisciplinary research directions can help further the real-world application of MLIPs to accurately model complex materials at device scale.
EGraFFBench: Evaluation of Equivariant Graph Neural Network Force Fields for Atomistic Simulations
Oct 03, 2023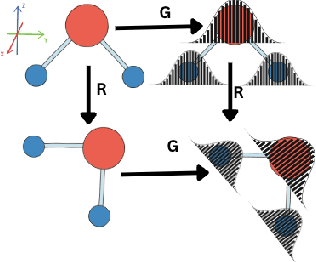
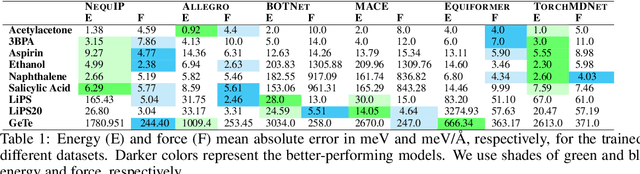
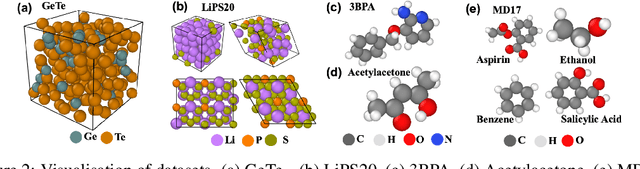
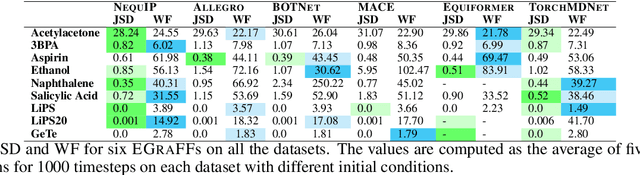
Equivariant graph neural networks force fields (EGraFFs) have shown great promise in modelling complex interactions in atomic systems by exploiting the graphs' inherent symmetries. Recent works have led to a surge in the development of novel architectures that incorporate equivariance-based inductive biases alongside architectural innovations like graph transformers and message passing to model atomic interactions. However, thorough evaluations of these deploying EGraFFs for the downstream task of real-world atomistic simulations, is lacking. To this end, here we perform a systematic benchmarking of 6 EGraFF algorithms (NequIP, Allegro, BOTNet, MACE, Equiformer, TorchMDNet), with the aim of understanding their capabilities and limitations for realistic atomistic simulations. In addition to our thorough evaluation and analysis on eight existing datasets based on the benchmarking literature, we release two new benchmark datasets, propose four new metrics, and three new challenging tasks. The new datasets and tasks evaluate the performance of EGraFF to out-of-distribution data, in terms of different crystal structures, temperatures, and new molecules. Interestingly, evaluation of the EGraFF models based on dynamic simulations reveals that having a lower error on energy or force does not guarantee stable or reliable simulation or faithful replication of the atomic structures. Moreover, we find that no model clearly outperforms other models on all datasets and tasks. Importantly, we show that the performance of all the models on out-of-distribution datasets is unreliable, pointing to the need for the development of a foundation model for force fields that can be used in real-world simulations. In summary, this work establishes a rigorous framework for evaluating machine learning force fields in the context of atomic simulations and points to open research challenges within this domain.