 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGFFMERGE: Efficient Merging of Graph Neural Force Fields and Beyond
Jun 02, 2026Graph Neural Networks (GNNs) have revolutionized Neural Force Fields for atomistic simulations, achieving near-quantum accuracy at reduced cost, yet adapting these models to new chemical systems requires expensive retraining of foundation models. Inspired by model merging in vision and language processing, we introduce GFFMERGE, the first principled framework for closed-form model merging in GNNs. We exploit the linear structure of message-passing layers and formulate merging as a convex embedding-alignment problem with an analytical solution. Through the first systematic benchmarking of model merging for GNNs, we show that existing methods designed for vision and language catastrophically fail on force field regression, while GFFMERGE recovers performance approaching gold standard joint training. Across molecular (MD17, MD22), solid-state (LiPS20), and large-scale graph benchmarks, GFFMERGE and GNNMERGE (its generic GNN counterpart) achieve 5-27$\times$ speedups while enabling modular composition of specialized models. Remarkably, our closed-form solution alone outperforms all baseline methods before fine-tuning and provides superior initialization for faster, data-efficient convergence.
A Multi-Plant Machine Learning Framework for Emission Prediction, Forecasting, and Control in Cement Manufacturing
Apr 21, 2026Cement production is among the largest contributors to industrial air pollution, emitting ~3 Mt NOx/year. The industry-standard mitigation approach, selective non-catalytic reduction (SNCR), exhibits low NH3 utilization efficiency, resulting in operational inefficiencies and increased reagent costs. Here, we develop a data-driven framework for emission control using large-scale operational data from four cement plants worldwide. Benchmarking nine machine learning architectures, we observe that prediction error varies ~3-5x across plants due to variation in data richness. Incorporating short-term process history nearly triples NOx prediction accuracy, revealing that NOx formation carries substantial process memory, a timescale dependence that is absent in CO and CO2. Further, we develop models that forecast NOx overshoots as early as nine minutes, providing a buffer for operational adjustments. The developed framework controls NOx formation at the source, reducing NH3 consumption in downstream SNCR. Surrogate model projections estimate a ~34-64% reduction in NOx while preserving clinker quality, corresponding to a reduction of ~290 t NOx/year and ~58,000 USD/year in NH3 savings. This work establishes a generalizable framework for data-driven emission control, offering a pathway toward low-emission operation without structural modifications or additional hardware, with potential applicability to other hard-to-abate industries such as steel, glass, and lime.
AI scientists produce results without reasoning scientifically
Apr 20, 2026Large language model (LLM)-based systems are increasingly deployed to conduct scientific research autonomously, yet whether their reasoning adheres to the epistemic norms that make scientific inquiry self-correcting is poorly understood. Here, we evaluate LLM-based scientific agents across eight domains, spanning workflow execution to hypothesis-driven inquiry, through more than 25,000 agent runs and two complementary lenses: (i) a systematic performance analysis that decomposes the contributions of the base model and the agent scaffold, and (ii) a behavioral analysis of the epistemological structure of agent reasoning. We observe that the base model is the primary determinant of both performance and behavior, accounting for 41.4% of explained variance versus 1.5% for the scaffold. Across all configurations, evidence is ignored in 68% of traces, refutation-driven belief revision occurs in 26%, and convergent multi-test evidence is rare. The same reasoning pattern appears whether the agent executes a computational workflow or conducts hypothesis-driven inquiry. They persist even when agents receive near-complete successful reasoning trajectories as context, and the resulting unreliability compounds across repeated trials in epistemically demanding domains. Thus, current LLM-based agents execute scientific workflows but do not exhibit the epistemic patterns that characterize scientific reasoning. Outcome-based evaluation cannot detect these failures, and scaffold engineering alone cannot repair them. Until reasoning itself becomes a training target, the scientific knowledge produced by such agents cannot be justified by the process that generated it.
Sustainable Materials Discovery in the Era of Artificial Intelligence
Jan 29, 2026Artificial intelligence (AI) has transformed materials discovery, enabling rapid exploration of chemical space through generative models and surrogate screening. Yet current AI workflows optimize performance first, deferring sustainability to post synthesis assessment. This creates inefficiency by the time environmental burdens are quantified, resources have been invested in potentially unsustainable solutions. The disconnect between atomic scale design and lifecycle assessment (LCA) reflects fundamental challenges, data scarcity across heterogeneous sources, scale gaps from atoms to industrial systems, uncertainty in synthesis pathways, and the absence of frameworks that co-optimize performance with environmental impact. We propose to integrate upstream machine learning (ML) assisted materials discovery with downstream lifecycle assessment into a uniform ML-LCA environment. The framework ML-LCA integrates five components, information extraction for building materials-environment knowledge bases, harmonized databases linking properties to sustainability metrics, multi-scale models bridging atomic properties to lifecycle impacts, ensemble prediction of manufacturing pathways with uncertainty quantification, and uncertainty-aware optimization enabling simultaneous performance-sustainability navigation. Case studies spanning glass, cement, semiconductor photoresists, and polymers demonstrate both necessity and feasibility while identifying material-specific integration challenges. Realizing ML-LCA demands coordinated advances in data infrastructure, ex-ante assessment methodologies, multi-objective optimization, and regulatory alignment enabling the discovery of materials that are sustainable by design rather than by chance.
On the Equivalence of Regression and Classification
Nov 06, 2025A formal link between regression and classification has been tenuous. Even though the margin maximization term $\|w\|$ is used in support vector regression, it has at best been justified as a regularizer. We show that a regression problem with $M$ samples lying on a hyperplane has a one-to-one equivalence with a linearly separable classification task with $2M$ samples. We show that margin maximization on the equivalent classification task leads to a different regression formulation than traditionally used. Using the equivalence, we demonstrate a ``regressability'' measure, that can be used to estimate the difficulty of regressing a dataset, without needing to first learn a model for it. We use the equivalence to train neural networks to learn a linearizing map, that transforms input variables into a space where a linear regressor is adequate.
Energy & Force Regression on DFT Trajectories is Not Enough for Universal Machine Learning Interatomic Potentials
Feb 05, 2025



Universal Machine Learning Interactomic Potentials (MLIPs) enable accelerated simulations for materials discovery. However, current research efforts fail to impactfully utilize MLIPs due to: 1. Overreliance on Density Functional Theory (DFT) for MLIP training data creation; 2. MLIPs' inability to reliably and accurately perform large-scale molecular dynamics (MD) simulations for diverse materials; 3. Limited understanding of MLIPs' underlying capabilities. To address these shortcomings, we aargue that MLIP research efforts should prioritize: 1. Employing more accurate simulation methods for large-scale MLIP training data creation (e.g. Coupled Cluster Theory) that cover a wide range of materials design spaces; 2. Creating MLIP metrology tools that leverage large-scale benchmarking, visualization, and interpretability analyses to provide a deeper understanding of MLIPs' inner workings; 3. Developing computationally efficient MLIPs to execute MD simulations that accurately model a broad set of materials properties. Together, these interdisciplinary research directions can help further the real-world application of MLIPs to accurately model complex materials at device scale.
CoNOAir: A Neural Operator for Forecasting Carbon Monoxide Evolution in Cities
Jan 13, 2025



Carbon Monoxide (CO) is a dominant pollutant in urban areas due to the energy generation from fossil fuels for industry, automobile, and domestic requirements. Forecasting the evolution of CO in real-time can enable the deployment of effective early warning systems and intervention strategies. However, the computational cost associated with the physics and chemistry-based simulation makes it prohibitive to implement such a model at the city and country scale. To address this challenge, here, we present a machine learning model based on neural operator, namely, Complex Neural Operator for Air Quality (CoNOAir), that can effectively forecast CO concentrations. We demonstrate this by developing a country-level model for short-term (hourly) and long-term (72-hour) forecasts of CO concentrations. Our model outperforms state-of-the-art models such as Fourier neural operators (FNO) and provides reliable predictions for both short and long-term forecasts. We further analyse the capability of the model to capture extreme events and generate forecasts in urban cities in India. Interestingly, we observe that the model predicts the next hour CO concentrations with R2 values greater than 0.95 for all the cities considered. The deployment of such a model can greatly assist the governing bodies to provide early warning, plan intervention strategies, and develop effective strategies by considering several what-if scenarios. Altogether, the present approach could provide a fillip to real-time predictions of CO pollution in urban cities.
Industrial-scale Prediction of Cement Clinker Phases using Machine Learning
Dec 16, 2024



Cement production, exceeding 4.1 billion tonnes and contributing 2.4 tonnes of CO2 annually, faces critical challenges in quality control and process optimization. While traditional process models for cement manufacturing are confined to steady-state conditions with limited predictive capability for mineralogical phases, modern plants operate under dynamic conditions that demand real-time quality assessment. Here, exploiting a comprehensive two-year operational dataset from an industrial cement plant, we present a machine learning framework that accurately predicts clinker mineralogy from process data. Our model achieves unprecedented prediction accuracy for major clinker phases while requiring minimal input parameters, demonstrating robust performance under varying operating conditions. Through post-hoc explainable algorithms, we interpret the hierarchical relationships between clinker oxides and phase formation, providing insights into the functioning of an otherwise black-box model. This digital twin framework can potentially enable real-time optimization of cement production, thereby providing a route toward reducing material waste and ensuring quality while reducing the associated emissions under real plant conditions. Our approach represents a significant advancement in industrial process control, offering a scalable solution for sustainable cement manufacturing.
Foundational Large Language Models for Materials Research
Dec 12, 2024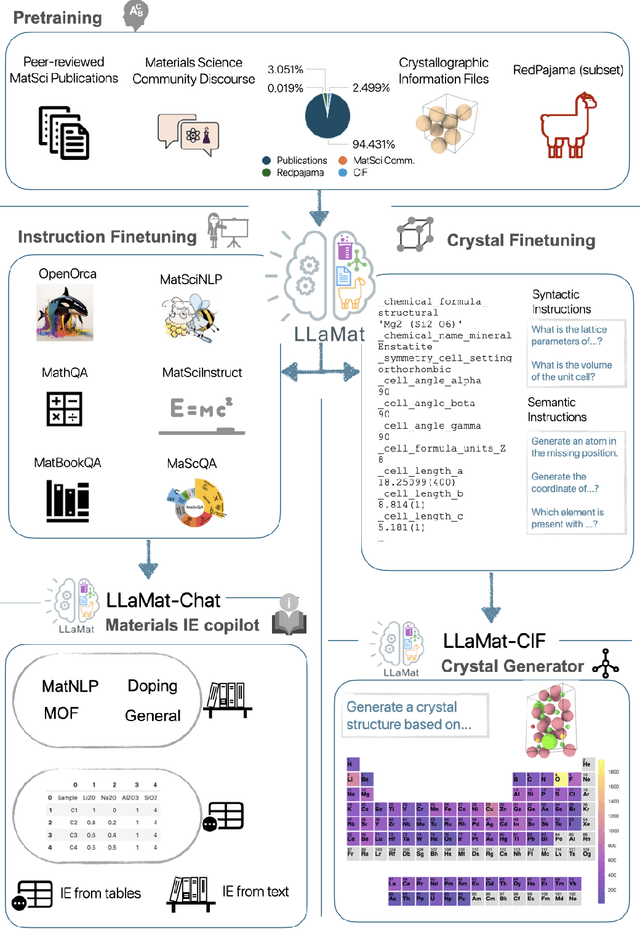
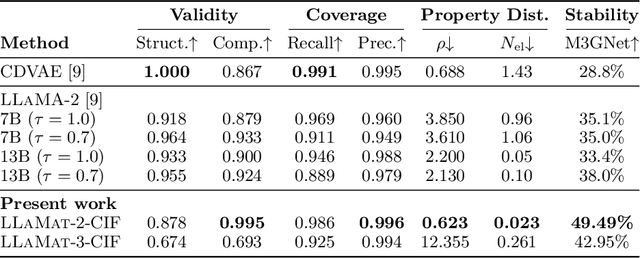
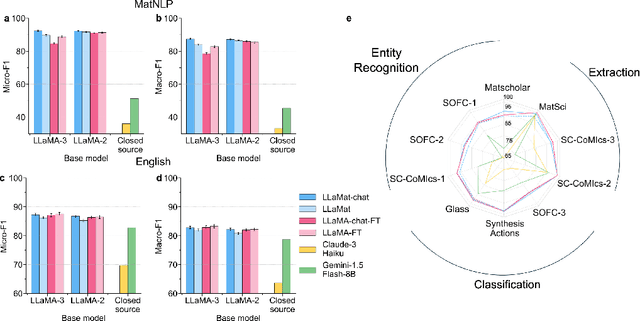
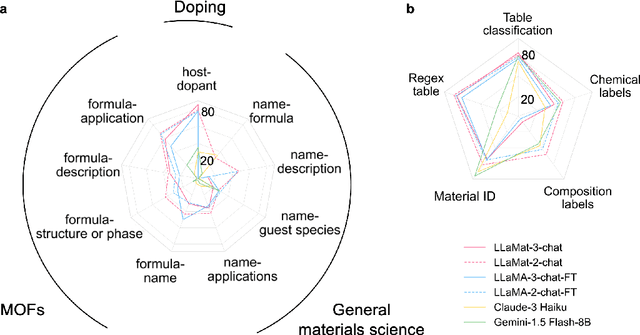
Materials discovery and development are critical for addressing global challenges. Yet, the exponential growth in materials science literature comprising vast amounts of textual data has created significant bottlenecks in knowledge extraction, synthesis, and scientific reasoning. Large Language Models (LLMs) offer unprecedented opportunities to accelerate materials research through automated analysis and prediction. Still, their effective deployment requires domain-specific adaptation for understanding and solving domain-relevant tasks. Here, we present LLaMat, a family of foundational models for materials science developed through continued pretraining of LLaMA models on an extensive corpus of materials literature and crystallographic data. Through systematic evaluation, we demonstrate that LLaMat excels in materials-specific NLP and structured information extraction while maintaining general linguistic capabilities. The specialized LLaMat-CIF variant demonstrates unprecedented capabilities in crystal structure generation, predicting stable crystals with high coverage across the periodic table. Intriguingly, despite LLaMA-3's superior performance in comparison to LLaMA-2, we observe that LLaMat-2 demonstrates unexpectedly enhanced domain-specific performance across diverse materials science tasks, including structured information extraction from text and tables, more particularly in crystal structure generation, a potential adaptation rigidity in overtrained LLMs. Altogether, the present work demonstrates the effectiveness of domain adaptation towards developing practically deployable LLM copilots for materials research. Beyond materials science, our findings reveal important considerations for domain adaptation of LLMs, such as model selection, training methodology, and domain-specific performance, which may influence the development of specialized scientific AI systems.
Probing the limitations of multimodal language models for chemistry and materials research
Nov 25, 2024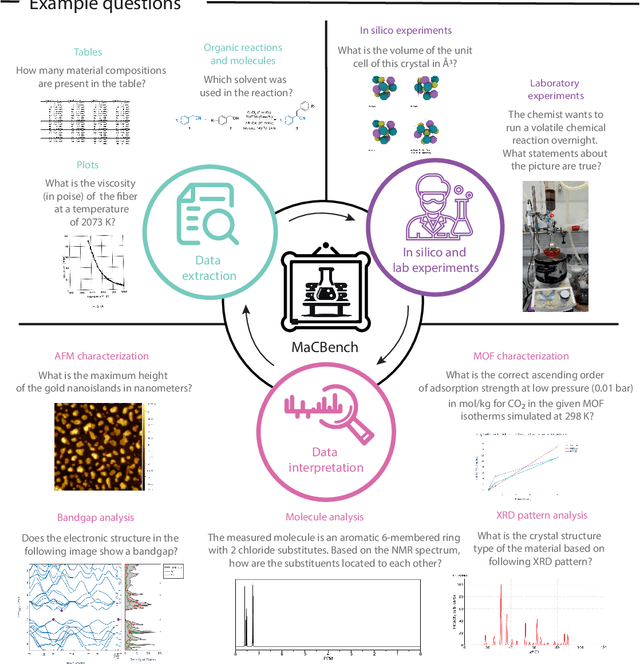
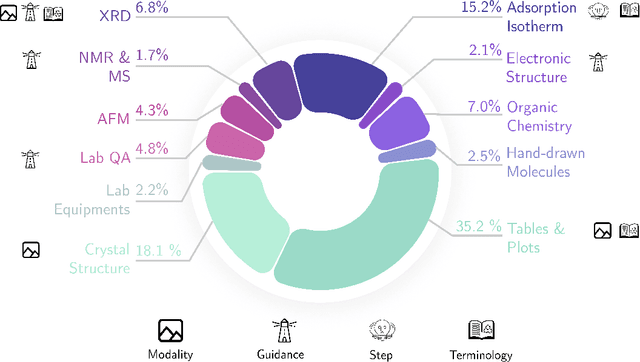
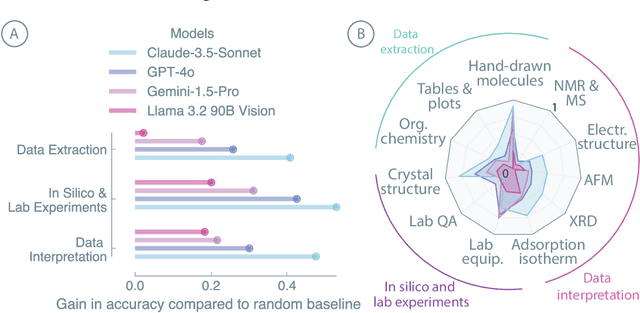
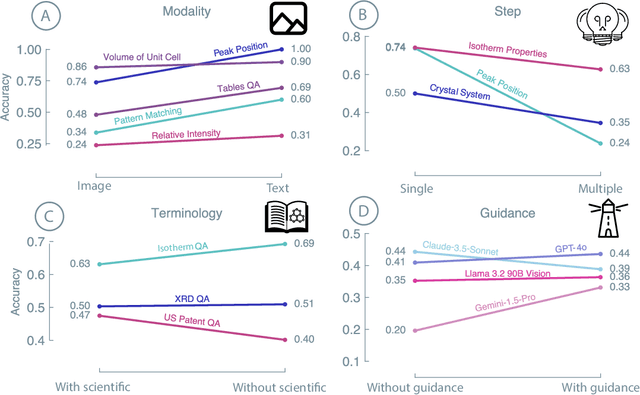
Recent advancements in artificial intelligence have sparked interest in scientific assistants that could support researchers across the full spectrum of scientific workflows, from literature review to experimental design and data analysis. A key capability for such systems is the ability to process and reason about scientific information in both visual and textual forms - from interpreting spectroscopic data to understanding laboratory setups. Here, we introduce MaCBench, a comprehensive benchmark for evaluating how vision-language models handle real-world chemistry and materials science tasks across three core aspects: data extraction, experimental understanding, and results interpretation. Through a systematic evaluation of leading models, we find that while these systems show promising capabilities in basic perception tasks - achieving near-perfect performance in equipment identification and standardized data extraction - they exhibit fundamental limitations in spatial reasoning, cross-modal information synthesis, and multi-step logical inference. Our insights have important implications beyond chemistry and materials science, suggesting that developing reliable multimodal AI scientific assistants may require advances in curating suitable training data and approaches to training those models.