 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStriderNET: A Graph Reinforcement Learning Approach to Optimize Atomic Structures on Rough Energy Landscapes
Paper and Code
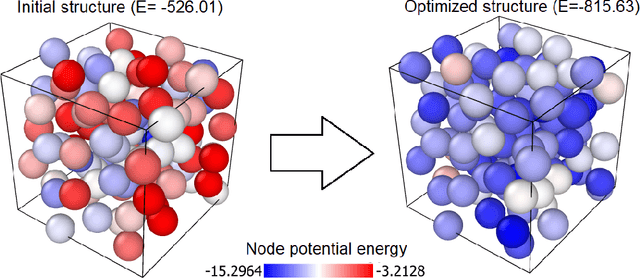
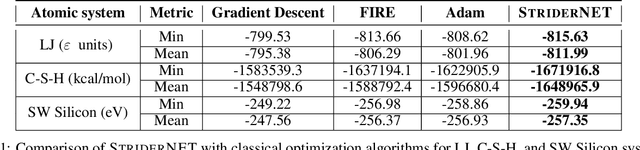
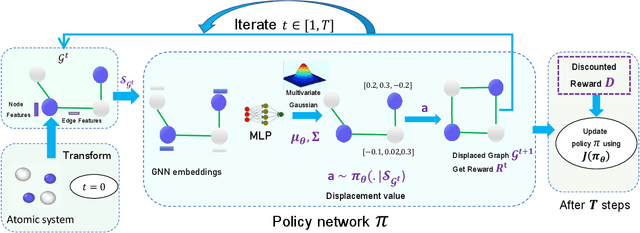

Optimization of atomic structures presents a challenging problem, due to their highly rough and non-convex energy landscape, with wide applications in the fields of drug design, materials discovery, and mechanics. Here, we present a graph reinforcement learning approach, StriderNET, that learns a policy to displace the atoms towards low energy configurations. We evaluate the performance of StriderNET on three complex atomic systems, namely, binary Lennard-Jones particles, calcium silicate hydrates gel, and disordered silicon. We show that StriderNET outperforms all classical optimization algorithms and enables the discovery of a lower energy minimum. In addition, StriderNET exhibits a higher rate of reaching minima with energies, as confirmed by the average over multiple realizations. Finally, we show that StriderNET exhibits inductivity to unseen system sizes that are an order of magnitude different from the training system.