 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStiefel Flow Matching for Moment-Constrained Structure Elucidation
Dec 17, 2024



Molecular structure elucidation is a fundamental step in understanding chemical phenomena, with applications in identifying molecules in natural products, lab syntheses, forensic samples, and the interstellar medium. We consider the task of predicting a molecule's all-atom 3D structure given only its molecular formula and moments of inertia, motivated by the ability of rotational spectroscopy to measure these moments. While existing generative models can conditionally sample 3D structures with approximately correct moments, this soft conditioning fails to leverage the many digits of precision afforded by experimental rotational spectroscopy. To address this, we first show that the space of $n$-atom point clouds with a fixed set of moments of inertia is embedded in the Stiefel manifold $\mathrm{St}(n, 4)$. We then propose Stiefel Flow Matching as a generative model for elucidating 3D structure under exact moment constraints. Additionally, we learn simpler and shorter flows by finding approximate solutions for equivariant optimal transport on the Stiefel manifold. Empirically, enforcing exact moment constraints allows Stiefel Flow Matching to achieve higher success rates and faster sampling than Euclidean diffusion models, even on high-dimensional manifolds corresponding to large molecules in the GEOM dataset.
Reflection-Equivariant Diffusion for 3D Structure Determination from Isotopologue Rotational Spectra in Natural Abundance
Oct 17, 2023



Structure determination is necessary to identify unknown organic molecules, such as those in natural products, forensic samples, the interstellar medium, and laboratory syntheses. Rotational spectroscopy enables structure determination by providing accurate 3D information about small organic molecules via their moments of inertia. Using these moments, Kraitchman analysis determines isotopic substitution coordinates, which are the unsigned $|x|,|y|,|z|$ coordinates of all atoms with natural isotopic abundance, including carbon, nitrogen, and oxygen. While unsigned substitution coordinates can verify guesses of structures, the missing $+/-$ signs make it challenging to determine the actual structure from the substitution coordinates alone. To tackle this inverse problem, we develop KREED (Kraitchman REflection-Equivariant Diffusion), a generative diffusion model that infers a molecule's complete 3D structure from its molecular formula, moments of inertia, and unsigned substitution coordinates of heavy atoms. KREED's top-1 predictions identify the correct 3D structure with >98% accuracy on the QM9 and GEOM datasets when provided with substitution coordinates of all heavy atoms with natural isotopic abundance. When substitution coordinates are restricted to only a subset of carbons, accuracy is retained at 91% on QM9 and 32% on GEOM. On a test set of experimentally measured substitution coordinates gathered from the literature, KREED predicts the correct all-atom 3D structure in 25 of 33 cases, demonstrating experimental applicability for context-free 3D structure determination with rotational spectroscopy.
Recent advances in the Self-Referencing Embedding Strings (SELFIES) library
Feb 07, 2023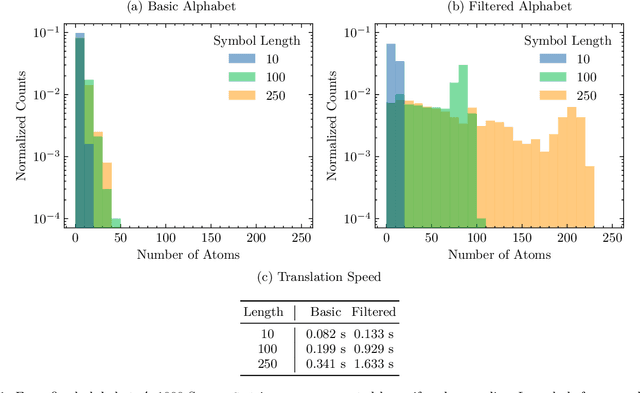
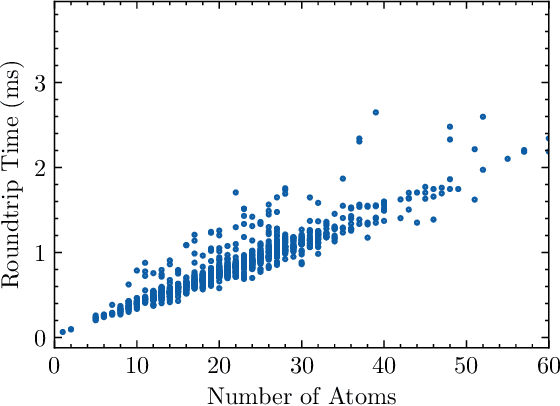

String-based molecular representations play a crucial role in cheminformatics applications, and with the growing success of deep learning in chemistry, have been readily adopted into machine learning pipelines. However, traditional string-based representations such as SMILES are often prone to syntactic and semantic errors when produced by generative models. To address these problems, a novel representation, SELF-referencIng Embedded Strings (SELFIES), was proposed that is inherently 100% robust, alongside an accompanying open-source implementation. Since then, we have generalized SELFIES to support a wider range of molecules and semantic constraints and streamlined its underlying grammar. We have implemented this updated representation in subsequent versions of \selfieslib, where we have also made major advances with respect to design, efficiency, and supported features. Hence, we present the current status of \selfieslib (version 2.1.1) in this manuscript.
If Influence Functions are the Answer, Then What is the Question?
Sep 12, 2022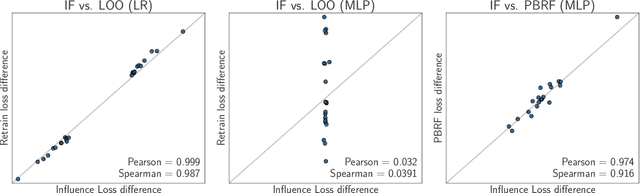


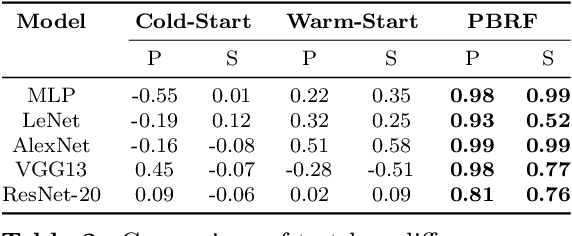
Influence functions efficiently estimate the effect of removing a single training data point on a model's learned parameters. While influence estimates align well with leave-one-out retraining for linear models, recent works have shown this alignment is often poor in neural networks. In this work, we investigate the specific factors that cause this discrepancy by decomposing it into five separate terms. We study the contributions of each term on a variety of architectures and datasets and how they vary with factors such as network width and training time. While practical influence function estimates may be a poor match to leave-one-out retraining for nonlinear networks, we show they are often a good approximation to a different object we term the proximal Bregman response function (PBRF). Since the PBRF can still be used to answer many of the questions motivating influence functions, such as identifying influential or mislabeled examples, our results suggest that current algorithms for influence function estimation give more informative results than previous error analyses would suggest.
SELFIES and the future of molecular string representations
Mar 31, 2022
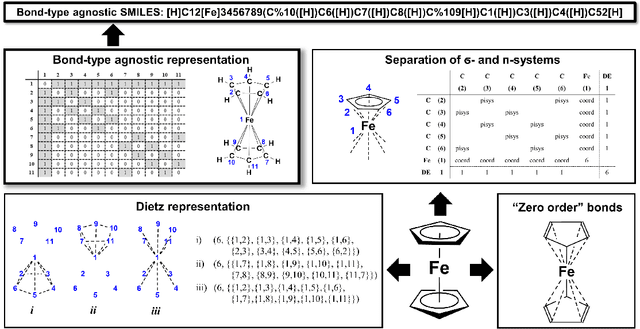
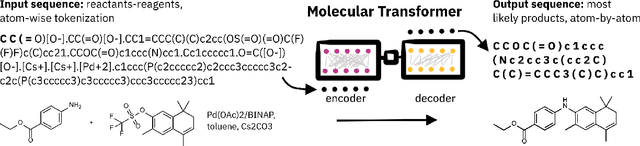
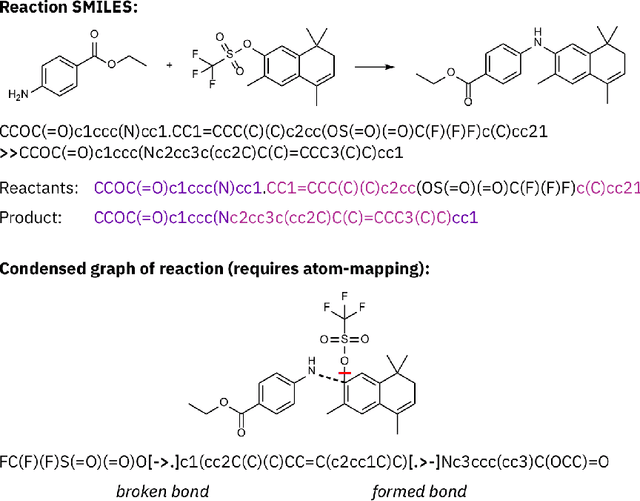
Artificial intelligence (AI) and machine learning (ML) are expanding in popularity for broad applications to challenging tasks in chemistry and materials science. Examples include the prediction of properties, the discovery of new reaction pathways, or the design of new molecules. The machine needs to read and write fluently in a chemical language for each of these tasks. Strings are a common tool to represent molecular graphs, and the most popular molecular string representation, SMILES, has powered cheminformatics since the late 1980s. However, in the context of AI and ML in chemistry, SMILES has several shortcomings -- most pertinently, most combinations of symbols lead to invalid results with no valid chemical interpretation. To overcome this issue, a new language for molecules was introduced in 2020 that guarantees 100\% robustness: SELFIES (SELF-referencIng Embedded Strings). SELFIES has since simplified and enabled numerous new applications in chemistry. In this manuscript, we look to the future and discuss molecular string representations, along with their respective opportunities and challenges. We propose 16 concrete Future Projects for robust molecular representations. These involve the extension toward new chemical domains, exciting questions at the interface of AI and robust languages and interpretability for both humans and machines. We hope that these proposals will inspire several follow-up works exploiting the full potential of molecular string representations for the future of AI in chemistry and materials science.