 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRobin: A multi-agent system for automating scientific discovery
May 19, 2025Scientific discovery is driven by the iterative process of background research, hypothesis generation, experimentation, and data analysis. Despite recent advancements in applying artificial intelligence to scientific discovery, no system has yet automated all of these stages in a single workflow. Here, we introduce Robin, the first multi-agent system capable of fully automating the key intellectual steps of the scientific process. By integrating literature search agents with data analysis agents, Robin can generate hypotheses, propose experiments, interpret experimental results, and generate updated hypotheses, achieving a semi-autonomous approach to scientific discovery. By applying this system, we were able to identify a novel treatment for dry age-related macular degeneration (dAMD), the major cause of blindness in the developed world. Robin proposed enhancing retinal pigment epithelium phagocytosis as a therapeutic strategy, and identified and validated a promising therapeutic candidate, ripasudil. Ripasudil is a clinically-used rho kinase (ROCK) inhibitor that has never previously been proposed for treating dAMD. To elucidate the mechanism of ripasudil-induced upregulation of phagocytosis, Robin then proposed and analyzed a follow-up RNA-seq experiment, which revealed upregulation of ABCA1, a critical lipid efflux pump and possible novel target. All hypotheses, experimental plans, data analyses, and data figures in the main text of this report were produced by Robin. As the first AI system to autonomously discover and validate a novel therapeutic candidate within an iterative lab-in-the-loop framework, Robin establishes a new paradigm for AI-driven scientific discovery.
MDCrow: Automating Molecular Dynamics Workflows with Large Language Models
Feb 13, 2025



Molecular dynamics (MD) simulations are essential for understanding biomolecular systems but remain challenging to automate. Recent advances in large language models (LLM) have demonstrated success in automating complex scientific tasks using LLM-based agents. In this paper, we introduce MDCrow, an agentic LLM assistant capable of automating MD workflows. MDCrow uses chain-of-thought over 40 expert-designed tools for handling and processing files, setting up simulations, analyzing the simulation outputs, and retrieving relevant information from literature and databases. We assess MDCrow's performance across 25 tasks of varying required subtasks and difficulty, and we evaluate the agent's robustness to both difficulty and prompt style. \texttt{gpt-4o} is able to complete complex tasks with low variance, followed closely by \texttt{llama3-405b}, a compelling open-source model. While prompt style does not influence the best models' performance, it has significant effects on smaller models.
Aviary: training language agents on challenging scientific tasks
Dec 30, 2024



Solving complex real-world tasks requires cycles of actions and observations. This is particularly true in science, where tasks require many cycles of analysis, tool use, and experimentation. Language agents are promising for automating intellectual tasks in science because they can interact with tools via natural language or code. Yet their flexibility creates conceptual and practical challenges for software implementations, since agents may comprise non-standard components such as internal reasoning, planning, tool usage, as well as the inherent stochasticity of temperature-sampled language models. Here, we introduce Aviary, an extensible gymnasium for language agents. We formalize agents as policies solving language-grounded partially observable Markov decision processes, which we term language decision processes. We then implement five environments, including three challenging scientific environments: (1) manipulating DNA constructs for molecular cloning, (2) answering research questions by accessing scientific literature, and (3) engineering protein stability. These environments were selected for their focus on multi-step reasoning and their relevance to contemporary biology research. Finally, with online training and scaling inference-time compute, we show that language agents backed by open-source, non-frontier LLMs can match and exceed both frontier LLM agents and human experts on multiple tasks at up to 100x lower inference cost.
LAB-Bench: Measuring Capabilities of Language Models for Biology Research
Jul 16, 2024There is widespread optimism that frontier Large Language Models (LLMs) and LLM-augmented systems have the potential to rapidly accelerate scientific discovery across disciplines. Today, many benchmarks exist to measure LLM knowledge and reasoning on textbook-style science questions, but few if any benchmarks are designed to evaluate language model performance on practical tasks required for scientific research, such as literature search, protocol planning, and data analysis. As a step toward building such benchmarks, we introduce the Language Agent Biology Benchmark (LAB-Bench), a broad dataset of over 2,400 multiple choice questions for evaluating AI systems on a range of practical biology research capabilities, including recall and reasoning over literature, interpretation of figures, access and navigation of databases, and comprehension and manipulation of DNA and protein sequences. Importantly, in contrast to previous scientific benchmarks, we expect that an AI system that can achieve consistently high scores on the more difficult LAB-Bench tasks would serve as a useful assistant for researchers in areas such as literature search and molecular cloning. As an initial assessment of the emergent scientific task capabilities of frontier language models, we measure performance of several against our benchmark and report results compared to human expert biology researchers. We will continue to update and expand LAB-Bench over time, and expect it to serve as a useful tool in the development of automated research systems going forward. A public subset of LAB-Bench is available for use at the following URL: https://huggingface.co/datasets/futurehouse/lab-bench
A Review of Large Language Models and Autonomous Agents in Chemistry
Jun 26, 2024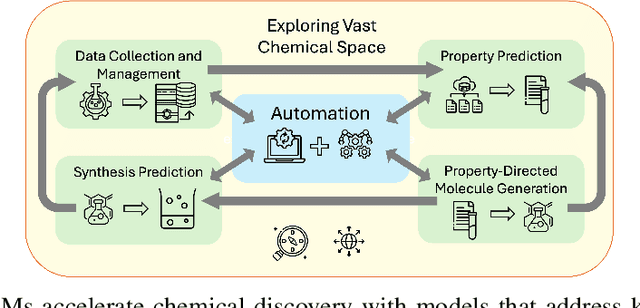

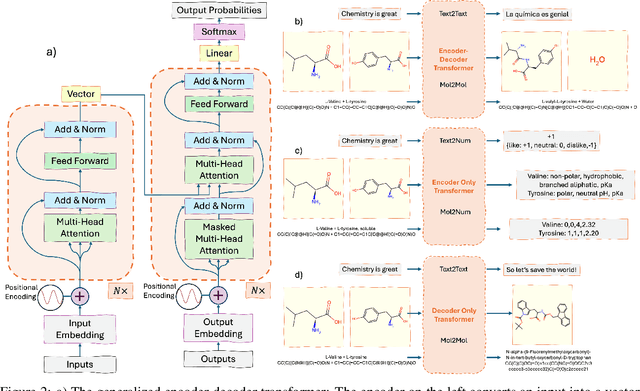
Large language models (LLMs) are emerging as a powerful tool in chemistry across multiple domains. In chemistry, LLMs are able to accurately predict properties, design new molecules, optimize synthesis pathways, and accelerate drug and material discovery. A core emerging idea is combining LLMs with chemistry-specific tools like synthesis planners and databases, leading to so-called "agents." This review covers LLMs' recent history, current capabilities, design, challenges specific to chemistry, and future directions. Particular attention is given to agents and their emergence as a cross-chemistry paradigm. Agents have proven effective in diverse domains of chemistry, but challenges remain. It is unclear if creating domain-specific versus generalist agents and developing autonomous pipelines versus "co-pilot" systems will accelerate chemistry. An emerging direction is the development of multi-agent systems using a human-in-the-loop approach. Due to the incredibly fast development of this field, a repository has been built to keep track of the latest studies: https://github.com/ur-whitelab/LLMs-in-science.
PaperQA: Retrieval-Augmented Generative Agent for Scientific Research
Dec 14, 2023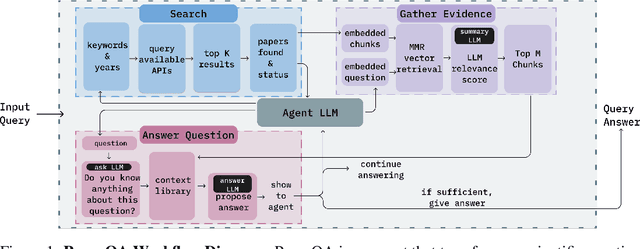



Large Language Models (LLMs) generalize well across language tasks, but suffer from hallucinations and uninterpretability, making it difficult to assess their accuracy without ground-truth. Retrieval-Augmented Generation (RAG) models have been proposed to reduce hallucinations and provide provenance for how an answer was generated. Applying such models to the scientific literature may enable large-scale, systematic processing of scientific knowledge. We present PaperQA, a RAG agent for answering questions over the scientific literature. PaperQA is an agent that performs information retrieval across full-text scientific articles, assesses the relevance of sources and passages, and uses RAG to provide answers. Viewing this agent as a question answering model, we find it exceeds performance of existing LLMs and LLM agents on current science QA benchmarks. To push the field closer to how humans perform research on scientific literature, we also introduce LitQA, a more complex benchmark that requires retrieval and synthesis of information from full-text scientific papers across the literature. Finally, we demonstrate PaperQA's matches expert human researchers on LitQA.
Predicting small molecules solubilities on endpoint devices using deep ensemble neural networks
Jul 11, 2023

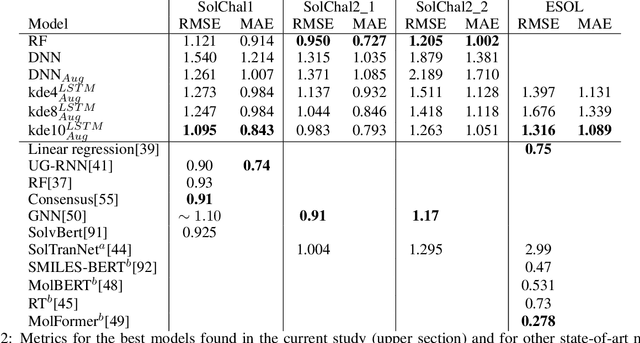

Aqueous solubility is a valuable yet challenging property to predict. Computing solubility using first-principles methods requires accounting for the competing effects of entropy and enthalpy, resulting in long computations for relatively poor accuracy. Data-driven approaches, such as deep learning, offer improved accuracy and computational efficiency but typically lack uncertainty quantification. Additionally, ease of use remains a concern for any computational technique, resulting in the sustained popularity of group-based contribution methods. In this work, we addressed these problems with a deep learning model with predictive uncertainty that runs on a static website (without a server). This approach moves computing needs onto the website visitor without requiring installation, removing the need to pay for and maintain servers. Our model achieves satisfactory results in solubility prediction. Furthermore, we demonstrate how to create molecular property prediction models that balance uncertainty and ease of use. The code is available at \url{https://github.com/ur-whitelab/mol.dev}, and the model is usable at \url{https://mol.dev}.
14 Examples of How LLMs Can Transform Materials Science and Chemistry: A Reflection on a Large Language Model Hackathon
Jun 13, 2023



Chemistry and materials science are complex. Recently, there have been great successes in addressing this complexity using data-driven or computational techniques. Yet, the necessity of input structured in very specific forms and the fact that there is an ever-growing number of tools creates usability and accessibility challenges. Coupled with the reality that much data in these disciplines is unstructured, the effectiveness of these tools is limited. Motivated by recent works that indicated that large language models (LLMs) might help address some of these issues, we organized a hackathon event on the applications of LLMs in chemistry, materials science, and beyond. This article chronicles the projects built as part of this hackathon. Participants employed LLMs for various applications, including predicting properties of molecules and materials, designing novel interfaces for tools, extracting knowledge from unstructured data, and developing new educational applications. The diverse topics and the fact that working prototypes could be generated in less than two days highlight that LLMs will profoundly impact the future of our fields. The rich collection of ideas and projects also indicates that the applications of LLMs are not limited to materials science and chemistry but offer potential benefits to a wide range of scientific disciplines.
Active Learning in Symbolic Regression with Physical Constraints
May 19, 2023Evolutionary symbolic regression (SR) fits a symbolic equation to data, which gives a concise interpretable model. We explore using SR as a method to propose which data to gather in an active learning setting with physical constraints. SR with active learning proposes which experiments to do next. Active learning is done with query by committee, where the Pareto frontier of equations is the committee. The physical constraints improve proposed equations in very low data settings. These approaches reduce the data required for SR and achieves state of the art results in data required to rediscover known equations.
Censoring chemical data to mitigate dual use risk
Apr 20, 2023The dual use of machine learning applications, where models can be used for both beneficial and malicious purposes, presents a significant challenge. This has recently become a particular concern in chemistry, where chemical datasets containing sensitive labels (e.g. toxicological information) could be used to develop predictive models that identify novel toxins or chemical warfare agents. To mitigate dual use risks, we propose a model-agnostic method of selectively noising datasets while preserving the utility of the data for training deep neural networks in a beneficial region. We evaluate the effectiveness of the proposed method across least squares, a multilayer perceptron, and a graph neural network. Our findings show selectively noised datasets can induce model variance and bias in predictions for sensitive labels with control, suggesting the safe sharing of datasets containing sensitive information is feasible. We also find omitting sensitive data often increases model variance sufficiently to mitigate dual use. This work is proposed as a foundation for future research on enabling more secure and collaborative data sharing practices and safer machine learning applications in chemistry.