 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEfficient Training of Boltzmann Generators Using Off-Policy Log-Dispersion Regularization
Feb 03, 2026Sampling from unnormalized probability densities is a central challenge in computational science. Boltzmann generators are generative models that enable independent sampling from the Boltzmann distribution of physical systems at a given temperature. However, their practical success depends on data-efficient training, as both simulation data and target energy evaluations are costly. To this end, we propose off-policy log-dispersion regularization (LDR), a novel regularization framework that builds on a generalization of the log-variance objective. We apply LDR in the off-policy setting in combination with standard data-based training objectives, without requiring additional on-policy samples. LDR acts as a shape regularizer of the energy landscape by leveraging additional information in the form of target energy labels. The proposed regularization framework is broadly applicable, supporting unbiased or biased simulation datasets as well as purely variational training without access to target samples. Across all benchmarks, LDR improves both final performance and data efficiency, with sample efficiency gains of up to one order of magnitude.
Graph Recognition via Subgraph Prediction
Jan 21, 2026Despite tremendous improvements in tasks such as image classification, object detection, and segmentation, the recognition of visual relationships, commonly modeled as the extraction of a graph from an image, remains a challenging task. We believe that this mainly stems from the fact that there is no canonical way to approach the visual graph recognition task. Most existing solutions are specific to a problem and cannot be transferred between different contexts out-of-the box, even though the conceptual problem remains the same. With broad applicability and simplicity in mind, in this paper we develop a method, \textbf{Gra}ph Recognition via \textbf{S}ubgraph \textbf{P}rediction (\textbf{GraSP}), for recognizing graphs in images. We show across several synthetic benchmarks and one real-world application that our method works with a set of diverse types of graphs and their drawings, and can be transferred between tasks without task-specific modifications, paving the way to a more unified framework for visual graph recognition.
Multi-stage Bayesian optimisation for dynamic decision-making in self-driving labs
Dec 17, 2025Self-driving laboratories (SDLs) are combining recent technological advances in robotics, automation, and machine learning based data analysis and decision-making to perform autonomous experimentation toward human-directed goals without requiring any direct human intervention. SDLs are successfully used in materials science, chemistry, and beyond, to optimise processes, materials, and devices in a systematic and data-efficient way. At present, the most widely used algorithm to identify the most informative next experiment is Bayesian optimisation. While relatively simple to apply to a wide range of optimisation problems, standard Bayesian optimisation relies on a fixed experimental workflow with a clear set of optimisation parameters and one or more measurable objective functions. This excludes the possibility of making on-the-fly decisions about changes in the planned sequence of operations and including intermediate measurements in the decision-making process. Therefore, many real-world experiments need to be adapted and simplified to be converted to the common setting in self-driving labs. In this paper, we introduce an extension to Bayesian optimisation that allows flexible sampling of multi-stage workflows and makes optimal decisions based on intermediate observables, which we call proxy measurements. We systematically compare the advantage of taking into account proxy measurements over conventional Bayesian optimisation, in which only the final measurement is observed. We find that over a wide range of scenarios, proxy measurements yield a substantial improvement, both in the time to find good solutions and in the overall optimality of found solutions. This not only paves the way to use more complex and thus more realistic experimental workflows in autonomous labs but also to smoothly combine simulations and experiments in the next generation of SDLs.
Learning Potential Energy Surfaces of Hydrogen Atom Transfer Reactions in Peptides
Aug 01, 2025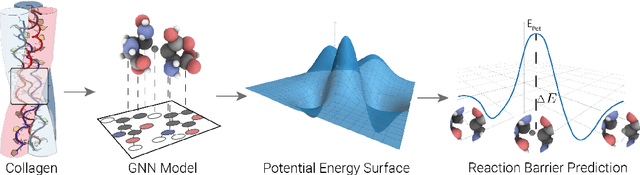
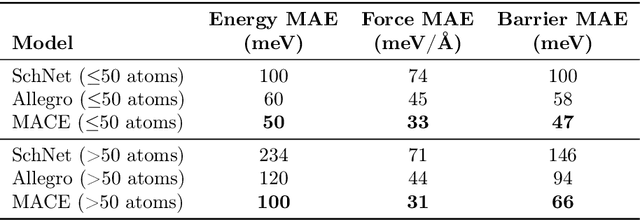
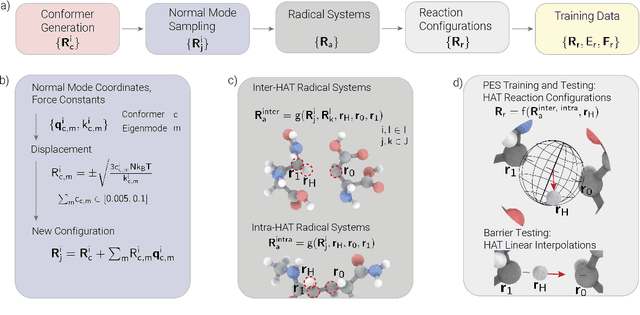

Hydrogen atom transfer (HAT) reactions are essential in many biological processes, such as radical migration in damaged proteins, but their mechanistic pathways remain incompletely understood. Simulating HAT is challenging due to the need for quantum chemical accuracy at biologically relevant scales; thus, neither classical force fields nor DFT-based molecular dynamics are applicable. Machine-learned potentials offer an alternative, able to learn potential energy surfaces (PESs) with near-quantum accuracy. However, training these models to generalize across diverse HAT configurations, especially at radical positions in proteins, requires tailored data generation and careful model selection. Here, we systematically generate HAT configurations in peptides to build large datasets using semiempirical methods and DFT. We benchmark three graph neural network architectures (SchNet, Allegro, and MACE) on their ability to learn HAT PESs and indirectly predict reaction barriers from energy predictions. MACE consistently outperforms the others in energy, force, and barrier prediction, achieving a mean absolute error of 1.13 kcal/mol on out-of-distribution DFT barrier predictions. This accuracy enables integration of ML potentials into large-scale collagen simulations to compute reaction rates from predicted barriers, advancing mechanistic understanding of HAT and radical migration in peptides. We analyze scaling laws, model transferability, and cost-performance trade-offs, and outline strategies for improvement by combining ML potentials with transition state search algorithms and active learning. Our approach is generalizable to other biomolecular systems, enabling quantum-accurate simulations of chemical reactivity in complex environments.
Improving Counterfactual Truthfulness for Molecular Property Prediction through Uncertainty Quantification
Apr 03, 2025Explainable AI (xAI) interventions aim to improve interpretability for complex black-box models, not only to improve user trust but also as a means to extract scientific insights from high-performing predictive systems. In molecular property prediction, counterfactual explanations offer a way to understand predictive behavior by highlighting which minimal perturbations in the input molecular structure cause the greatest deviation in the predicted property. However, such explanations only allow for meaningful scientific insights if they reflect the distribution of the true underlying property -- a feature we define as counterfactual truthfulness. To increase this truthfulness, we propose the integration of uncertainty estimation techniques to filter counterfactual candidates with high predicted uncertainty. Through computational experiments with synthetic and real-world datasets, we demonstrate that traditional uncertainty estimation methods, such as ensembles and mean-variance estimation, can already substantially reduce the average prediction error and increase counterfactual truthfulness, especially for out-of-distribution settings. Our results highlight the importance and potential impact of incorporating uncertainty estimation into explainability methods, especially considering the relatively high effectiveness of low-effort interventions like model ensembles.
opXRD: Open Experimental Powder X-ray Diffraction Database
Mar 07, 2025Powder X-ray diffraction (pXRD) experiments are a cornerstone for materials structure characterization. Despite their widespread application, analyzing pXRD diffractograms still presents a significant challenge to automation and a bottleneck in high-throughput discovery in self-driving labs. Machine learning promises to resolve this bottleneck by enabling automated powder diffraction analysis. A notable difficulty in applying machine learning to this domain is the lack of sufficiently sized experimental datasets, which has constrained researchers to train primarily on simulated data. However, models trained on simulated pXRD patterns showed limited generalization to experimental patterns, particularly for low-quality experimental patterns with high noise levels and elevated backgrounds. With the Open Experimental Powder X-Ray Diffraction Database (opXRD), we provide an openly available and easily accessible dataset of labeled and unlabeled experimental powder diffractograms. Labeled opXRD data can be used to evaluate the performance of models on experimental data and unlabeled opXRD data can help improve the performance of models on experimental data, e.g. through transfer learning methods. We collected \numpatterns diffractograms, 2179 of them labeled, from a wide spectrum of materials classes. We hope this ongoing effort can guide machine learning research toward fully automated analysis of pXRD data and thus enable future self-driving materials labs.
Symmetry-Aware Bayesian Flow Networks for Crystal Generation
Feb 05, 2025The discovery of new crystalline materials is essential to scientific and technological progress. However, traditional trial-and-error approaches are inefficient due to the vast search space. Recent advancements in machine learning have enabled generative models to predict new stable materials by incorporating structural symmetries and to condition the generation on desired properties. In this work, we introduce SymmBFN, a novel symmetry-aware Bayesian Flow Network (BFN) for crystalline material generation that accurately reproduces the distribution of space groups found in experimentally observed crystals. SymmBFN substantially improves efficiency, generating stable structures at least 50 times faster than the next-best method. Furthermore, we demonstrate its capability for property-conditioned generation, enabling the design of materials with tailored properties. Our findings establish BFNs as an effective tool for accelerating the discovery of crystalline materials.
Temperature-Annealed Boltzmann Generators
Jan 31, 2025



Efficient sampling of unnormalized probability densities such as the Boltzmann distribution of molecular systems is a longstanding challenge. Next to conventional approaches like molecular dynamics or Markov chain Monte Carlo, variational approaches, such as training normalizing flows with the reverse Kullback-Leibler divergence, have been introduced. However, such methods are prone to mode collapse and often do not learn to sample the full configurational space. Here, we present temperature-annealed Boltzmann generators (TA-BG) to address this challenge. First, we demonstrate that training a normalizing flow with the reverse Kullback-Leibler divergence at high temperatures is possible without mode collapse. Furthermore, we introduce a reweighting-based training objective to anneal the distribution to lower target temperatures. We apply this methodology to three molecular systems of increasing complexity and, compared to the baseline, achieve better results in almost all metrics while requiring up to three times fewer target energy evaluations. For the largest system, our approach is the only method that accurately resolves the metastable states of the system.
PAL -- Parallel active learning for machine-learned potentials
Nov 30, 2024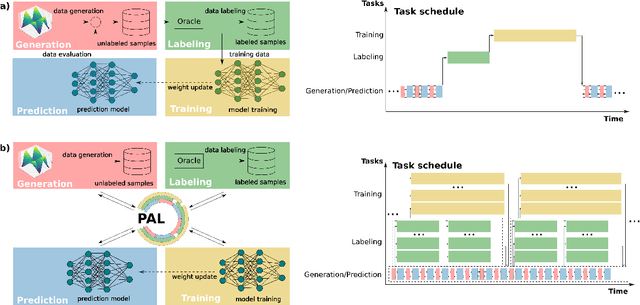

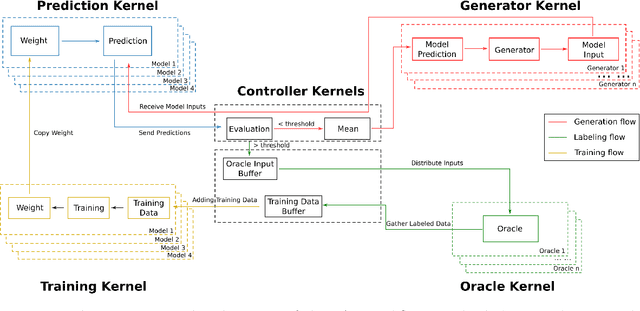
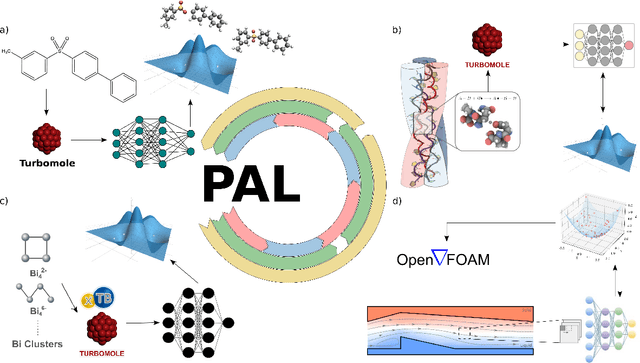
Constructing datasets representative of the target domain is essential for training effective machine learning models. Active learning (AL) is a promising method that iteratively extends training data to enhance model performance while minimizing data acquisition costs. However, current AL workflows often require human intervention and lack parallelism, leading to inefficiencies and underutilization of modern computational resources. In this work, we introduce PAL, an automated, modular, and parallel active learning library that integrates AL tasks and manages their execution and communication on shared- and distributed-memory systems using the Message Passing Interface (MPI). PAL provides users with the flexibility to design and customize all components of their active learning scenarios, including machine learning models with uncertainty estimation, oracles for ground truth labeling, and strategies for exploring the target space. We demonstrate that PAL significantly reduces computational overhead and improves scalability, achieving substantial speed-ups through asynchronous parallelization on CPU and GPU hardware. Applications of PAL to several real-world scenarios - including ground-state reactions in biomolecular systems, excited-state dynamics of molecules, simulations of inorganic clusters, and thermo-fluid dynamics - illustrate its effectiveness in accelerating the development of machine learning models. Our results show that PAL enables efficient utilization of high-performance computing resources in active learning workflows, fostering advancements in scientific research and engineering applications.
Global Concept Explanations for Graphs by Contrastive Learning
Apr 25, 2024



Beyond improving trust and validating model fairness, xAI practices also have the potential to recover valuable scientific insights in application domains where little to no prior human intuition exists. To that end, we propose a method to extract global concept explanations from the predictions of graph neural networks to develop a deeper understanding of the tasks underlying structure-property relationships. We identify concept explanations as dense clusters in the self-explaining Megan models subgraph latent space. For each concept, we optimize a representative prototype graph and optionally use GPT-4 to provide hypotheses about why each structure has a certain effect on the prediction. We conduct computational experiments on synthetic and real-world graph property prediction tasks. For the synthetic tasks we find that our method correctly reproduces the structural rules by which they were created. For real-world molecular property regression and classification tasks, we find that our method rediscovers established rules of thumb. More specifically, our results for molecular mutagenicity prediction indicate more fine-grained resolution of structural details than existing explainability methods, consistent with previous results from chemistry literature. Overall, our results show promising capability to extract the underlying structure-property relationships for complex graph property prediction tasks.