 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeConditional Normalizing Flows for Active Learning of Coarse-Grained Molecular Representations
Paper and Code
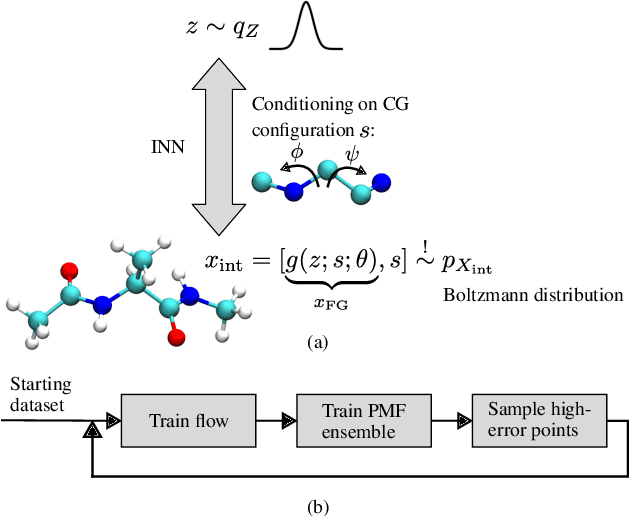

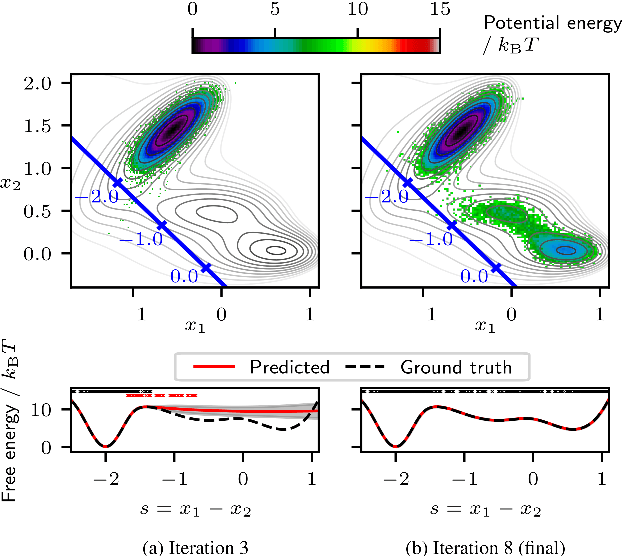
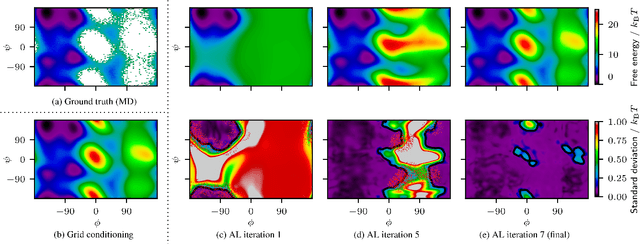
Efficient sampling of the Boltzmann distribution of molecular systems is a long-standing challenge. Recently, instead of generating long molecular dynamics simulations, generative machine learning methods such as normalizing flows have been used to learn the Boltzmann distribution directly, without samples. However, this approach is susceptible to mode collapse and thus often does not explore the full configurational space. In this work, we address this challenge by separating the problem into two levels, the fine-grained and coarse-grained degrees of freedom. A normalizing flow conditioned on the coarse-grained space yields a probabilistic connection between the two levels. To explore the configurational space, we employ coarse-grained simulations with active learning which allows us to update the flow and make all-atom potential energy evaluations only when necessary. Using alanine dipeptide as an example, we show that our methods obtain a speedup to molecular dynamics simulations of approximately 15.9 to 216.2 compared to the speedup of 4.5 of the current state-of-the-art machine learning approach.