 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMitigating Molecular Aggregation in Drug Discovery with Predictive Insights from Explainable AI
Paper and Code
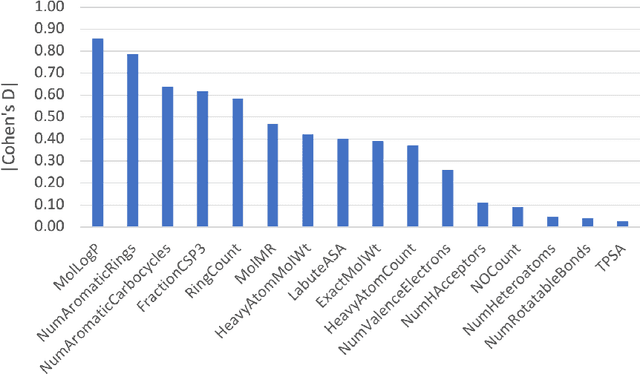
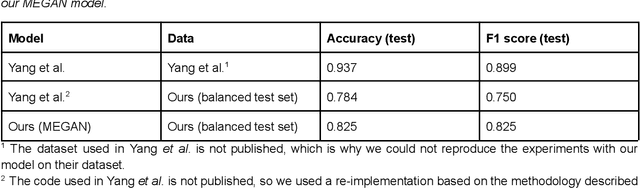
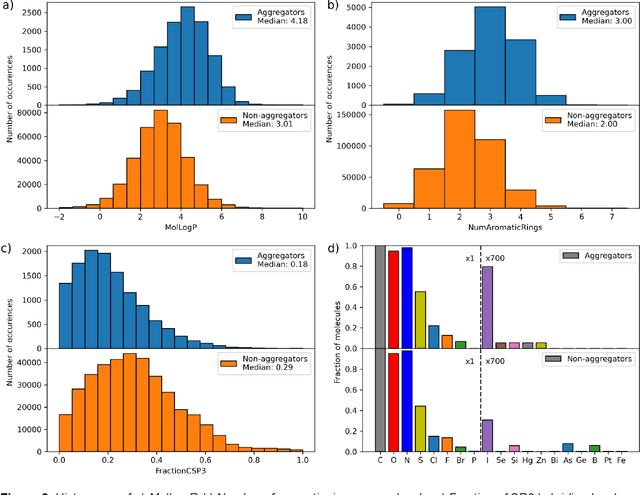
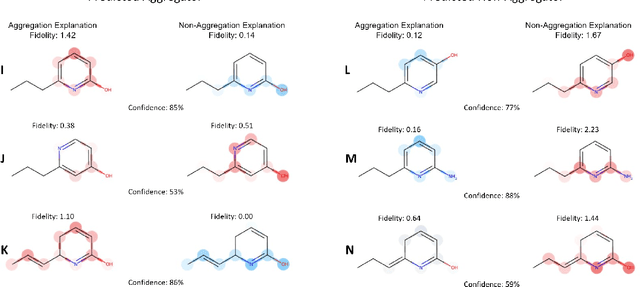
As the importance of high-throughput screening (HTS) continues to grow due to its value in early stage drug discovery and data generation for training machine learning models, there is a growing need for robust methods for pre-screening compounds to identify and prevent false-positive hits. Small, colloidally aggregating molecules are one of the primary sources of false-positive hits in high-throughput screens, making them an ideal candidate to target for removal from libraries using predictive pre-screening tools. However, a lack of understanding of the causes of molecular aggregation introduces difficulty in the development of predictive tools for detecting aggregating molecules. Herein, we present an examination of the molecular features differentiating datasets of aggregating and non-aggregating molecules, as well as a machine learning approach to predicting molecular aggregation. Our method uses explainable graph neural networks and counterfactuals to reliably predict and explain aggregation, giving additional insights and design rules for future screening. The integration of this method in HTS approaches will help combat false positives, providing better lead molecules more rapidly and thus accelerating drug discovery cycles.