 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImproving Counterfactual Truthfulness for Molecular Property Prediction through Uncertainty Quantification
Apr 03, 2025Explainable AI (xAI) interventions aim to improve interpretability for complex black-box models, not only to improve user trust but also as a means to extract scientific insights from high-performing predictive systems. In molecular property prediction, counterfactual explanations offer a way to understand predictive behavior by highlighting which minimal perturbations in the input molecular structure cause the greatest deviation in the predicted property. However, such explanations only allow for meaningful scientific insights if they reflect the distribution of the true underlying property -- a feature we define as counterfactual truthfulness. To increase this truthfulness, we propose the integration of uncertainty estimation techniques to filter counterfactual candidates with high predicted uncertainty. Through computational experiments with synthetic and real-world datasets, we demonstrate that traditional uncertainty estimation methods, such as ensembles and mean-variance estimation, can already substantially reduce the average prediction error and increase counterfactual truthfulness, especially for out-of-distribution settings. Our results highlight the importance and potential impact of incorporating uncertainty estimation into explainability methods, especially considering the relatively high effectiveness of low-effort interventions like model ensembles.
opXRD: Open Experimental Powder X-ray Diffraction Database
Mar 07, 2025Powder X-ray diffraction (pXRD) experiments are a cornerstone for materials structure characterization. Despite their widespread application, analyzing pXRD diffractograms still presents a significant challenge to automation and a bottleneck in high-throughput discovery in self-driving labs. Machine learning promises to resolve this bottleneck by enabling automated powder diffraction analysis. A notable difficulty in applying machine learning to this domain is the lack of sufficiently sized experimental datasets, which has constrained researchers to train primarily on simulated data. However, models trained on simulated pXRD patterns showed limited generalization to experimental patterns, particularly for low-quality experimental patterns with high noise levels and elevated backgrounds. With the Open Experimental Powder X-Ray Diffraction Database (opXRD), we provide an openly available and easily accessible dataset of labeled and unlabeled experimental powder diffractograms. Labeled opXRD data can be used to evaluate the performance of models on experimental data and unlabeled opXRD data can help improve the performance of models on experimental data, e.g. through transfer learning methods. We collected \numpatterns diffractograms, 2179 of them labeled, from a wide spectrum of materials classes. We hope this ongoing effort can guide machine learning research toward fully automated analysis of pXRD data and thus enable future self-driving materials labs.
Global Concept Explanations for Graphs by Contrastive Learning
Apr 25, 2024



Beyond improving trust and validating model fairness, xAI practices also have the potential to recover valuable scientific insights in application domains where little to no prior human intuition exists. To that end, we propose a method to extract global concept explanations from the predictions of graph neural networks to develop a deeper understanding of the tasks underlying structure-property relationships. We identify concept explanations as dense clusters in the self-explaining Megan models subgraph latent space. For each concept, we optimize a representative prototype graph and optionally use GPT-4 to provide hypotheses about why each structure has a certain effect on the prediction. We conduct computational experiments on synthetic and real-world graph property prediction tasks. For the synthetic tasks we find that our method correctly reproduces the structural rules by which they were created. For real-world molecular property regression and classification tasks, we find that our method rediscovers established rules of thumb. More specifically, our results for molecular mutagenicity prediction indicate more fine-grained resolution of structural details than existing explainability methods, consistent with previous results from chemistry literature. Overall, our results show promising capability to extract the underlying structure-property relationships for complex graph property prediction tasks.
Mitigating Molecular Aggregation in Drug Discovery with Predictive Insights from Explainable AI
Jun 03, 2023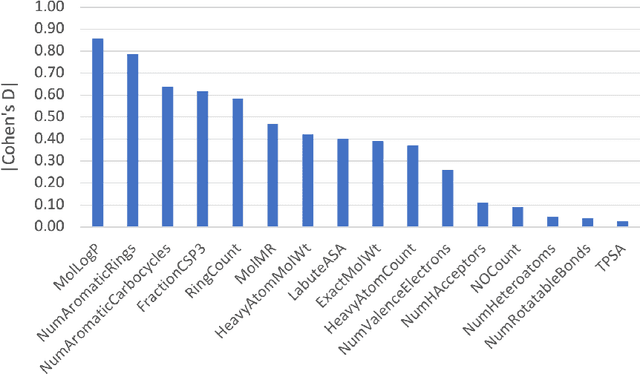
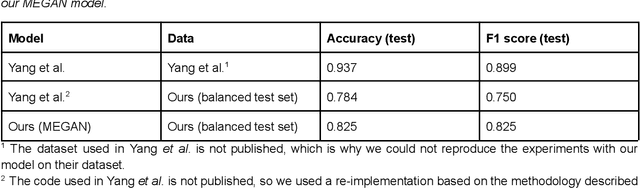
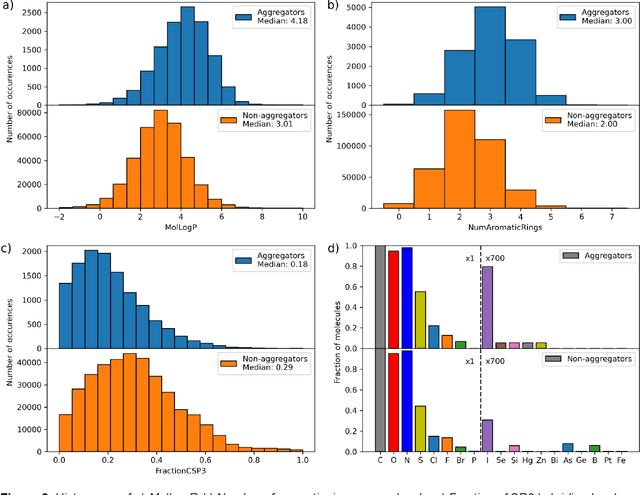
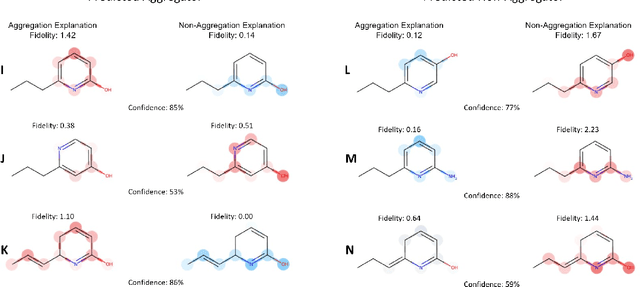
As the importance of high-throughput screening (HTS) continues to grow due to its value in early stage drug discovery and data generation for training machine learning models, there is a growing need for robust methods for pre-screening compounds to identify and prevent false-positive hits. Small, colloidally aggregating molecules are one of the primary sources of false-positive hits in high-throughput screens, making them an ideal candidate to target for removal from libraries using predictive pre-screening tools. However, a lack of understanding of the causes of molecular aggregation introduces difficulty in the development of predictive tools for detecting aggregating molecules. Herein, we present an examination of the molecular features differentiating datasets of aggregating and non-aggregating molecules, as well as a machine learning approach to predicting molecular aggregation. Our method uses explainable graph neural networks and counterfactuals to reliably predict and explain aggregation, giving additional insights and design rules for future screening. The integration of this method in HTS approaches will help combat false positives, providing better lead molecules more rapidly and thus accelerating drug discovery cycles.
Quantifying the Intrinsic Usefulness of Attributional Explanations for Graph Neural Networks with Artificial Simulatability Studies
May 25, 2023Despite the increasing relevance of explainable AI, assessing the quality of explanations remains a challenging issue. Due to the high costs associated with human-subject experiments, various proxy metrics are often used to approximately quantify explanation quality. Generally, one possible interpretation of the quality of an explanation is its inherent value for teaching a related concept to a student. In this work, we extend artificial simulatability studies to the domain of graph neural networks. Instead of costly human trials, we use explanation-supervisable graph neural networks to perform simulatability studies to quantify the inherent usefulness of attributional graph explanations. We perform an extensive ablation study to investigate the conditions under which the proposed analyses are most meaningful. We additionally validate our methods applicability on real-world graph classification and regression datasets. We find that relevant explanations can significantly boost the sample efficiency of graph neural networks and analyze the robustness towards noise and bias in the explanations. We believe that the notion of usefulness obtained from our proposed simulatability analysis provides a dimension of explanation quality that is largely orthogonal to the common practice of faithfulness and has great potential to expand the toolbox of explanation quality assessments, specifically for graph explanations.
MEGAN: Multi-Explanation Graph Attention Network
Nov 23, 2022Explainable artificial intelligence (XAI) methods are expected to improve trust during human-AI interactions, provide tools for model analysis and extend human understanding of complex problems. Explanation-supervised training allows to improve explanation quality by training self-explaining XAI models on ground truth or human-generated explanations. However, existing explanation methods have limited expressiveness and interoperability due to the fact that only single explanations in form of node and edge importance are generated. To that end we propose the novel multi-explanation graph attention network (MEGAN). Our fully differentiable, attention-based model features multiple explanation channels, which can be chosen independently of the task specifications. We first validate our model on a synthetic graph regression dataset. We show that for the special single explanation case, our model significantly outperforms existing post-hoc and explanation-supervised baseline methods. Furthermore, we demonstrate significant advantages when using two explanations, both in quantitative explanation measures as well as in human interpretability. Finally, we demonstrate our model's capabilities on multiple real-world datasets. We find that our model produces sparse high-fidelity explanations consistent with human intuition about those tasks and at the same time matches state-of-the-art graph neural networks in predictive performance, indicating that explanations and accuracy are not necessarily a trade-off.