 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning Potential Energy Surfaces of Hydrogen Atom Transfer Reactions in Peptides
Aug 01, 2025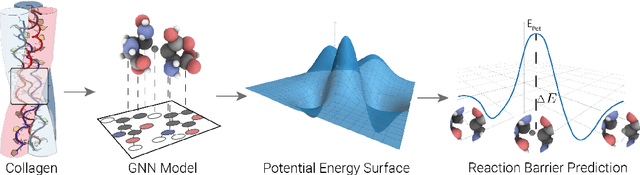
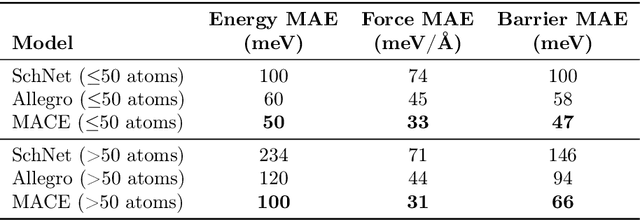
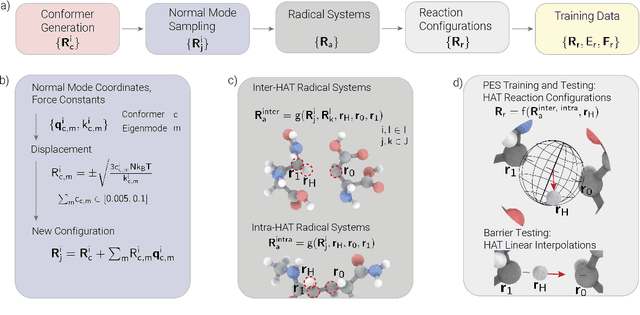

Hydrogen atom transfer (HAT) reactions are essential in many biological processes, such as radical migration in damaged proteins, but their mechanistic pathways remain incompletely understood. Simulating HAT is challenging due to the need for quantum chemical accuracy at biologically relevant scales; thus, neither classical force fields nor DFT-based molecular dynamics are applicable. Machine-learned potentials offer an alternative, able to learn potential energy surfaces (PESs) with near-quantum accuracy. However, training these models to generalize across diverse HAT configurations, especially at radical positions in proteins, requires tailored data generation and careful model selection. Here, we systematically generate HAT configurations in peptides to build large datasets using semiempirical methods and DFT. We benchmark three graph neural network architectures (SchNet, Allegro, and MACE) on their ability to learn HAT PESs and indirectly predict reaction barriers from energy predictions. MACE consistently outperforms the others in energy, force, and barrier prediction, achieving a mean absolute error of 1.13 kcal/mol on out-of-distribution DFT barrier predictions. This accuracy enables integration of ML potentials into large-scale collagen simulations to compute reaction rates from predicted barriers, advancing mechanistic understanding of HAT and radical migration in peptides. We analyze scaling laws, model transferability, and cost-performance trade-offs, and outline strategies for improvement by combining ML potentials with transition state search algorithms and active learning. Our approach is generalizable to other biomolecular systems, enabling quantum-accurate simulations of chemical reactivity in complex environments.
Neural networks trained on synthetically generated crystals can extract structural information from ICSD powder X-ray diffractograms
Mar 24, 2023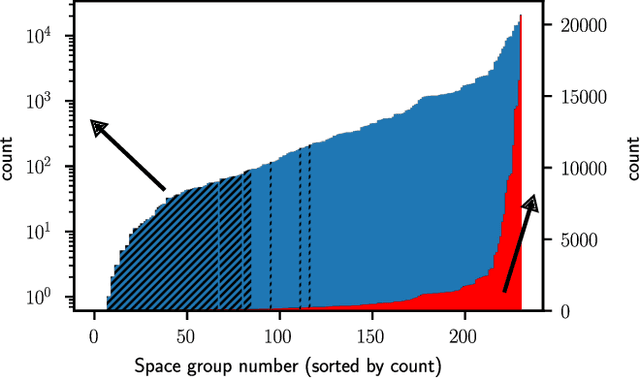
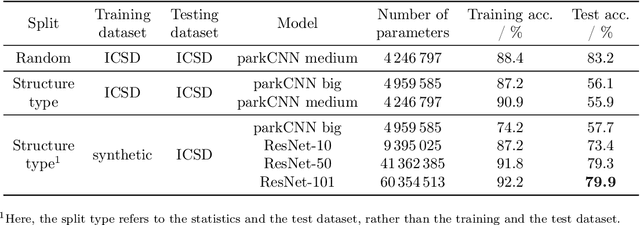
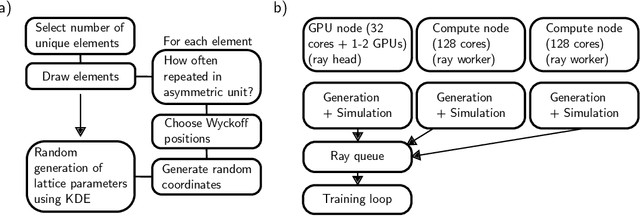
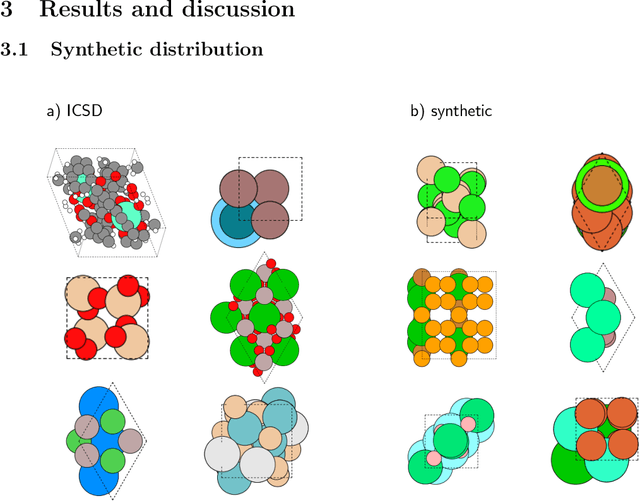
Machine learning techniques have successfully been used to extract structural information such as the crystal space group from powder X-ray diffractograms. However, training directly on simulated diffractograms from databases such as the ICSD is challenging due to its limited size, class-inhomogeneity, and bias toward certain structure types. We propose an alternative approach of generating synthetic crystals with random coordinates by using the symmetry operations of each space group. Based on this approach, we demonstrate online training of deep ResNet-like models on up to a few million unique on-the-fly generated synthetic diffractograms per hour. For our chosen task of space group classification, we achieved a test accuracy of 79.9% on unseen ICSD structure types from most space groups. This surpasses the 56.1% accuracy of the current state-of-the-art approach of training on ICSD crystals directly. Our results demonstrate that synthetically generated crystals can be used to extract structural information from ICSD powder diffractograms, which makes it possible to apply very large state-of-the-art machine learning models in the area of powder X-ray diffraction. We further show first steps toward applying our methodology to experimental data, where automated XRD data analysis is crucial, especially in high-throughput settings. While we focused on the prediction of the space group, our approach has the potential to be extended to related tasks in the future.
Connectivity Optimized Nested Graph Networks for Crystal Structures
Feb 27, 2023Graph neural networks (GNNs) have been applied to a large variety of applications in materials science and chemistry. Here, we recapitulate the graph construction for crystalline (periodic) materials and investigate its impact on the GNNs model performance. We suggest the asymmetric unit cell as a representation to reduce the number of atoms by using all symmetries of the system. With a simple but systematically built GNN architecture based on message passing and line graph templates, we furthermore introduce a general architecture (Nested Graph Network, NGN) that is applicable to a wide range of tasks and systematically improves state-of-the-art results on the MatBench benchmark datasets.
MEGAN: Multi-Explanation Graph Attention Network
Nov 23, 2022Explainable artificial intelligence (XAI) methods are expected to improve trust during human-AI interactions, provide tools for model analysis and extend human understanding of complex problems. Explanation-supervised training allows to improve explanation quality by training self-explaining XAI models on ground truth or human-generated explanations. However, existing explanation methods have limited expressiveness and interoperability due to the fact that only single explanations in form of node and edge importance are generated. To that end we propose the novel multi-explanation graph attention network (MEGAN). Our fully differentiable, attention-based model features multiple explanation channels, which can be chosen independently of the task specifications. We first validate our model on a synthetic graph regression dataset. We show that for the special single explanation case, our model significantly outperforms existing post-hoc and explanation-supervised baseline methods. Furthermore, we demonstrate significant advantages when using two explanations, both in quantitative explanation measures as well as in human interpretability. Finally, we demonstrate our model's capabilities on multiple real-world datasets. We find that our model produces sparse high-fidelity explanations consistent with human intuition about those tasks and at the same time matches state-of-the-art graph neural networks in predictive performance, indicating that explanations and accuracy are not necessarily a trade-off.
Implementing graph neural networks with TensorFlow-Keras
Mar 07, 2021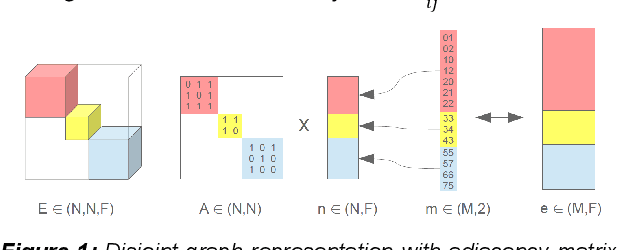
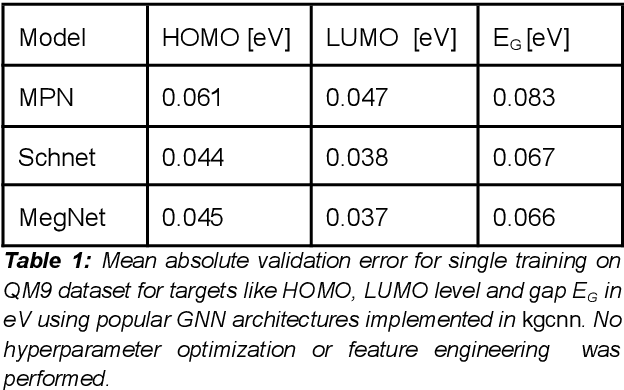
Graph neural networks are a versatile machine learning architecture that received a lot of attention recently. In this technical report, we present an implementation of convolution and pooling layers for TensorFlow-Keras models, which allows a seamless and flexible integration into standard Keras layers to set up graph models in a functional way. This implies the usage of mini-batches as the first tensor dimension, which can be realized via the new RaggedTensor class of TensorFlow best suited for graphs. We developed the Keras Graph Convolutional Neural Network Python package kgcnn based on TensorFlow-Keras that provides a set of Keras layers for graph networks which focus on a transparent tensor structure passed between layers and an ease-of-use mindset.
Analyzing dynamical disorder for charge transport in organic semiconductors via machine learning
Feb 02, 2021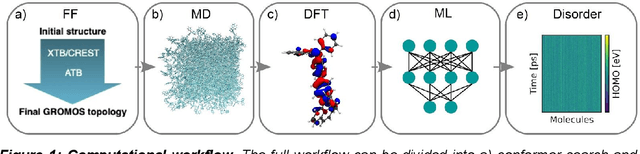
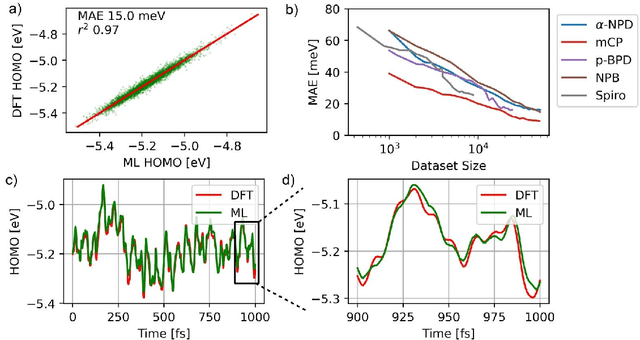
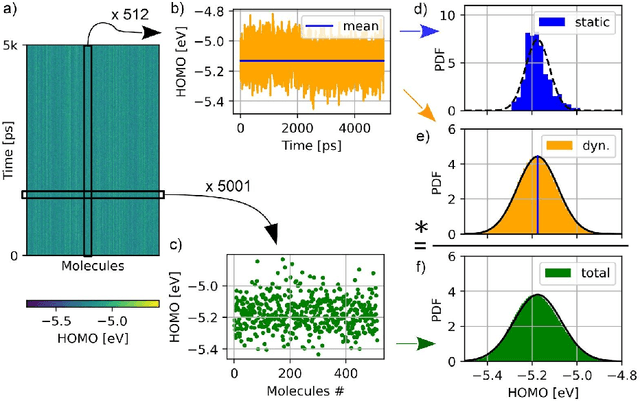
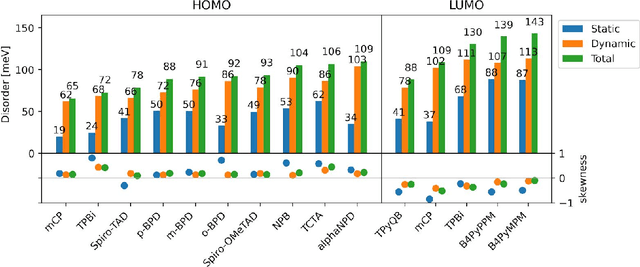
Organic semiconductors are indispensable for today's display technologies in form of organic light emitting diodes (OLEDs) and further optoelectronic applications. However, organic materials do not reach the same charge carrier mobility as inorganic semiconductors, limiting the efficiency of devices. To find or even design new organic semiconductors with higher charge carrier mobility, computational approaches, in particular multiscale models, are becoming increasingly important. However, such models are computationally very costly, especially when large systems and long time scales are required, which is the case to compute static and dynamic energy disorder, i.e. dominant factor to determine charge transport. Here we overcome this drawback by integrating machine learning models into multiscale simulations. This allows us to obtain unprecedented insight into relevant microscopic materials properties, in particular static and dynamic disorder contributions for a series of application-relevant molecules. We find that static disorder and thus the distribution of shallow traps is highly asymmetrical for many materials, impacting widely considered Gaussian disorder models. We furthermore analyse characteristic energy level fluctuation times and compare them to typical hopping rates to evaluate the importance of dynamic disorder for charge transport. We hope that our findings will significantly improve the accuracy of computational methods used to predict application relevant materials properties of organic semiconductors, and thus make these methods applicable for virtual materials design.