 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStrain Problems got you in a Twist? Try StrainRelief: A Quantum-Accurate Tool for Ligand Strain Calculations
Mar 17, 2025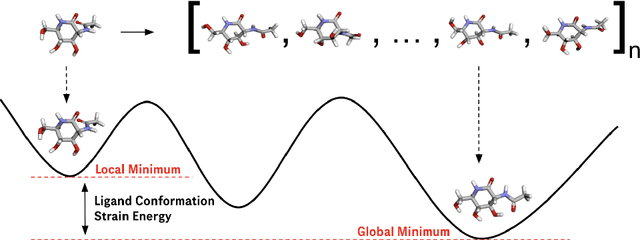
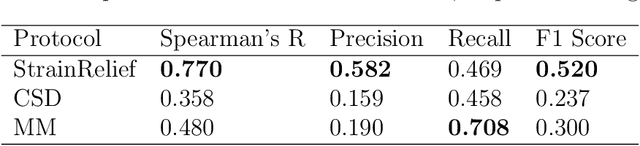


Ligand strain energy, the energy difference between the bound and unbound conformations of a ligand, is an important component of structure-based small molecule drug design. A large majority of observed ligands in protein-small molecule co-crystal structures bind in low-strain conformations, making strain energy a useful filter for structure-based drug design. In this work we present a tool for calculating ligand strain with a high accuracy. StrainRelief uses a MACE Neural Network Potential (NNP), trained on a large database of Density Functional Theory (DFT) calculations to estimate ligand strain of neutral molecules with quantum accuracy. We show that this tool estimates strain energy differences relative to DFT to within 1.4 kcal/mol, more accurately than alternative NNPs. These results highlight the utility of NNPs in drug discovery, and provide a useful tool for drug discovery teams.
Concept Bottleneck Language Models For protein design
Nov 09, 2024



We introduce Concept Bottleneck Protein Language Models (CB-pLM), a generative masked language model with a layer where each neuron corresponds to an interpretable concept. Our architecture offers three key benefits: i) Control: We can intervene on concept values to precisely control the properties of generated proteins, achieving a 3 times larger change in desired concept values compared to baselines. ii) Interpretability: A linear mapping between concept values and predicted tokens allows transparent analysis of the model's decision-making process. iii) Debugging: This transparency facilitates easy debugging of trained models. Our models achieve pre-training perplexity and downstream task performance comparable to traditional masked protein language models, demonstrating that interpretability does not compromise performance. While adaptable to any language model, we focus on masked protein language models due to their importance in drug discovery and the ability to validate our model's capabilities through real-world experiments and expert knowledge. We scale our CB-pLM from 24 million to 3 billion parameters, making them the largest Concept Bottleneck Models trained and the first capable of generative language modeling.
LLMs are Highly-Constrained Biophysical Sequence Optimizers
Oct 29, 2024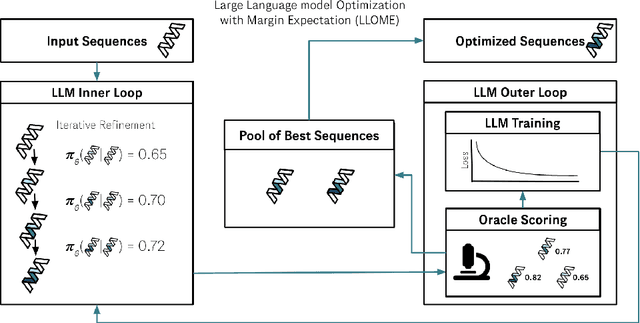
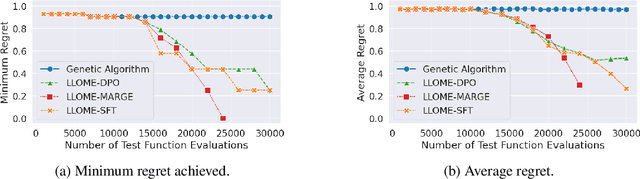
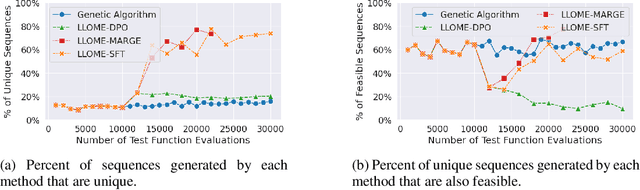
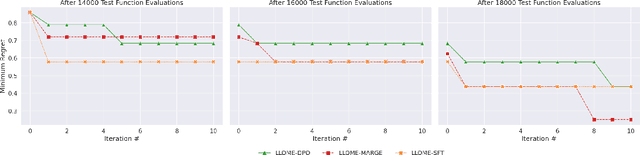
Large language models (LLMs) have recently shown significant potential in various biological tasks such as protein engineering and molecule design. These tasks typically involve black-box discrete sequence optimization, where the challenge lies in generating sequences that are not only biologically feasible but also adhere to hard fine-grained constraints. However, LLMs often struggle with such constraints, especially in biological contexts where verifying candidate solutions is costly and time-consuming. In this study, we explore the possibility of employing LLMs as highly-constrained bilevel optimizers through a methodology we refer to as Language Model Optimization with Margin Expectation (LLOME). This approach combines both offline and online optimization, utilizing limited oracle evaluations to iteratively enhance the sequences generated by the LLM. We additionally propose a novel training objective -- Margin-Aligned Expectation (MargE) -- that trains the LLM to smoothly interpolate between the reward and reference distributions. Lastly, we introduce a synthetic test suite that bears strong geometric similarity to real biophysical problems and enables rapid evaluation of LLM optimizers without time-consuming lab validation. Our findings reveal that, in comparison to genetic algorithm baselines, LLMs achieve significantly lower regret solutions while requiring fewer test function evaluations. However, we also observe that LLMs exhibit moderate miscalibration, are susceptible to generator collapse, and have difficulty finding the optimal solution when no explicit ground truth rewards are available.
Protein Discovery with Discrete Walk-Jump Sampling
Jun 08, 2023



We resolve difficulties in training and sampling from a discrete generative model by learning a smoothed energy function, sampling from the smoothed data manifold with Langevin Markov chain Monte Carlo (MCMC), and projecting back to the true data manifold with one-step denoising. Our Discrete Walk-Jump Sampling formalism combines the maximum likelihood training of an energy-based model and improved sample quality of a score-based model, while simplifying training and sampling by requiring only a single noise level. We evaluate the robustness of our approach on generative modeling of antibody proteins and introduce the distributional conformity score to benchmark protein generative models. By optimizing and sampling from our models for the proposed distributional conformity score, 97-100% of generated samples are successfully expressed and purified and 35% of functional designs show equal or improved binding affinity compared to known functional antibodies on the first attempt in a single round of laboratory experiments. We also report the first demonstration of long-run fast-mixing MCMC chains where diverse antibody protein classes are visited in a single MCMC chain.
Protein Design with Guided Discrete Diffusion
May 31, 2023A popular approach to protein design is to combine a generative model with a discriminative model for conditional sampling. The generative model samples plausible sequences while the discriminative model guides a search for sequences with high fitness. Given its broad success in conditional sampling, classifier-guided diffusion modeling is a promising foundation for protein design, leading many to develop guided diffusion models for structure with inverse folding to recover sequences. In this work, we propose diffusioN Optimized Sampling (NOS), a guidance method for discrete diffusion models that follows gradients in the hidden states of the denoising network. NOS makes it possible to perform design directly in sequence space, circumventing significant limitations of structure-based methods, including scarce data and challenging inverse design. Moreover, we use NOS to generalize LaMBO, a Bayesian optimization procedure for sequence design that facilitates multiple objectives and edit-based constraints. The resulting method, LaMBO-2, enables discrete diffusions and stronger performance with limited edits through a novel application of saliency maps. We apply LaMBO-2 to a real-world protein design task, optimizing antibodies for higher expression yield and binding affinity to a therapeutic target under locality and liability constraints, with 97% expression rate and 25% binding rate in exploratory in vitro experiments.
SupSiam: Non-contrastive Auxiliary Loss for Learning from Molecular Conformers
Feb 15, 2023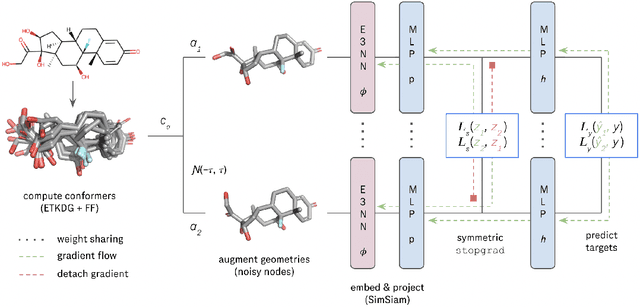

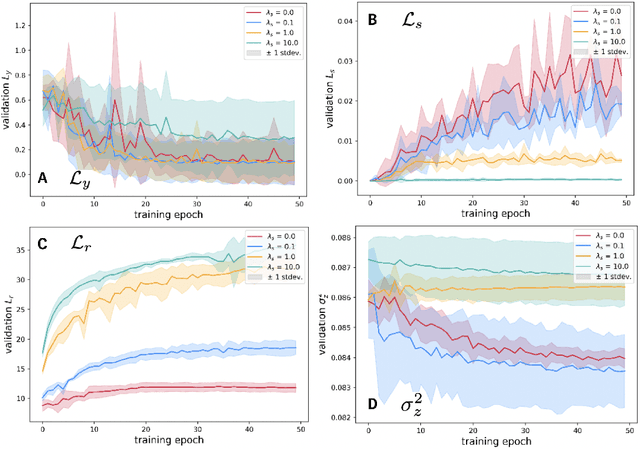

We investigate Siamese networks for learning related embeddings for augmented samples of molecular conformers. We find that a non-contrastive (positive-pair only) auxiliary task aids in supervised training of Euclidean neural networks (E3NNs) and increases manifold smoothness (MS) around point-cloud geometries. We demonstrate this property for multiple drug-activity prediction tasks while maintaining relevant performance metrics, and propose an extension of MS to probabilistic and regression settings. We provide an analysis of representation collapse, finding substantial effects of task-weighting, latent dimension, and regularization. We expect the presented protocol to aid in the development of reliable E3NNs from molecular conformers, even for small-data drug discovery programs.
A Green(er) World for A.I
Jan 27, 2023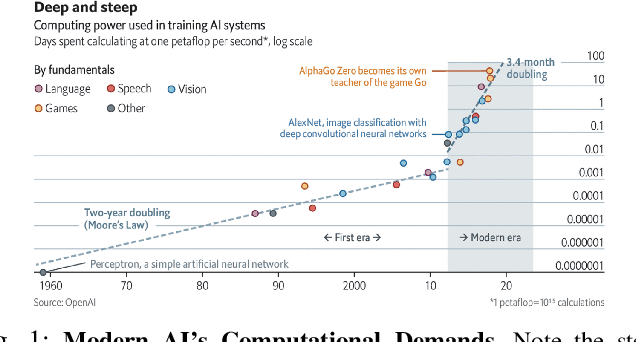
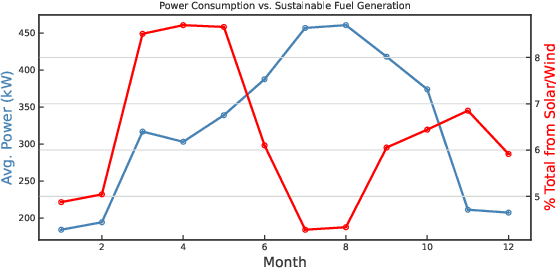
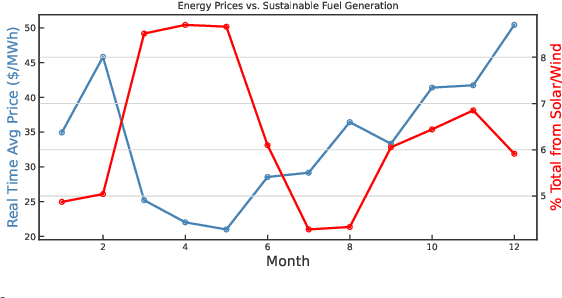
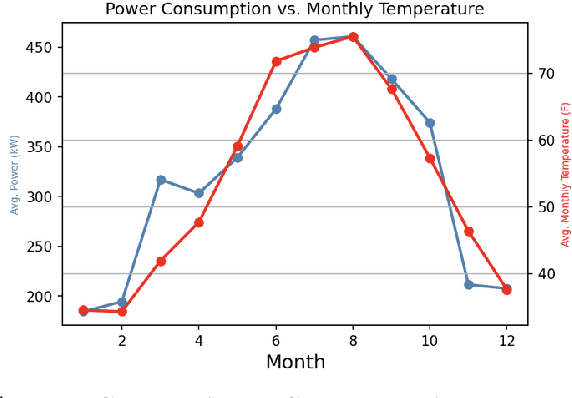
As research and practice in artificial intelligence (A.I.) grow in leaps and bounds, the resources necessary to sustain and support their operations also grow at an increasing pace. While innovations and applications from A.I. have brought significant advances, from applications to vision and natural language to improvements to fields like medical imaging and materials engineering, their costs should not be neglected. As we embrace a world with ever-increasing amounts of data as well as research and development of A.I. applications, we are sure to face an ever-mounting energy footprint to sustain these computational budgets, data storage needs, and more. But, is this sustainable and, more importantly, what kind of setting is best positioned to nurture such sustainable A.I. in both research and practice? In this paper, we outline our outlook for Green A.I. -- a more sustainable, energy-efficient and energy-aware ecosystem for developing A.I. across the research, computing, and practitioner communities alike -- and the steps required to arrive there. We present a bird's eye view of various areas for potential changes and improvements from the ground floor of AI's operational and hardware optimizations for datacenters/HPCs to the current incentive structures in the world of A.I. research and practice, and more. We hope these points will spur further discussion, and action, on some of these issues and their potential solutions.
* 8 pages, published in 2022 IEEE International Parallel and Distributed Processing Symposium Workshops (IPDPSW)
Graph Contrastive Learning for Materials
Nov 24, 2022


Recent work has shown the potential of graph neural networks to efficiently predict material properties, enabling high-throughput screening of materials. Training these models, however, often requires large quantities of labelled data, obtained via costly methods such as ab initio calculations or experimental evaluation. By leveraging a series of material-specific transformations, we introduce CrystalCLR, a framework for constrastive learning of representations with crystal graph neural networks. With the addition of a novel loss function, our framework is able to learn representations competitive with engineered fingerprinting methods. We also demonstrate that via model finetuning, contrastive pretraining can improve the performance of graph neural networks for prediction of material properties and significantly outperform traditional ML models that use engineered fingerprints. Lastly, we observe that CrystalCLR produces material representations that form clusters by compound class.
A Pareto-optimal compositional energy-based model for sampling and optimization of protein sequences
Oct 19, 2022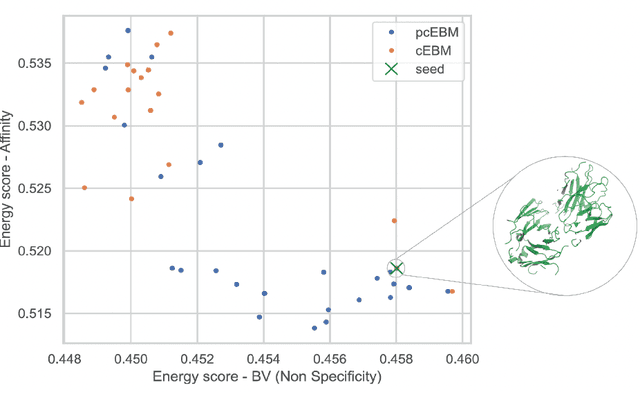
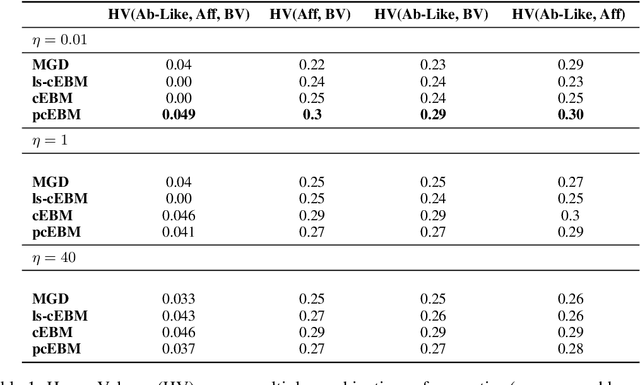
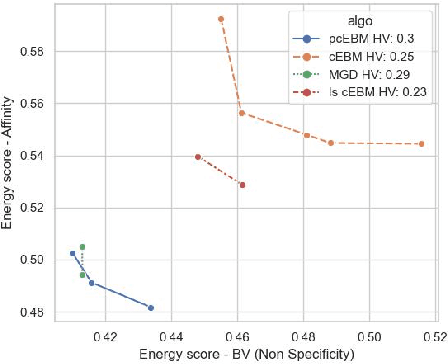
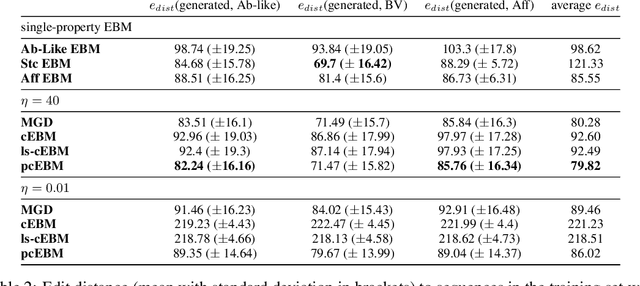
Deep generative models have emerged as a popular machine learning-based approach for inverse design problems in the life sciences. However, these problems often require sampling new designs that satisfy multiple properties of interest in addition to learning the data distribution. This multi-objective optimization becomes more challenging when properties are independent or orthogonal to each other. In this work, we propose a Pareto-compositional energy-based model (pcEBM), a framework that uses multiple gradient descent for sampling new designs that adhere to various constraints in optimizing distinct properties. We demonstrate its ability to learn non-convex Pareto fronts and generate sequences that simultaneously satisfy multiple desired properties across a series of real-world antibody design tasks.
SELFIES and the future of molecular string representations
Mar 31, 2022
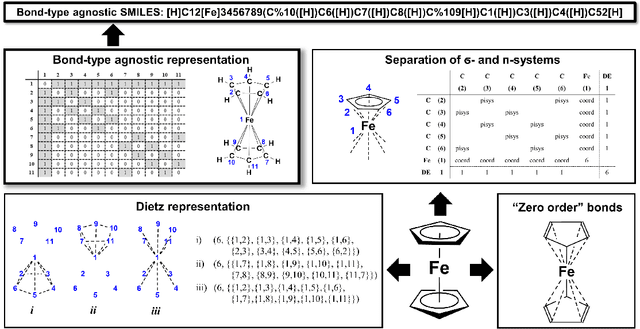
Artificial intelligence (AI) and machine learning (ML) are expanding in popularity for broad applications to challenging tasks in chemistry and materials science. Examples include the prediction of properties, the discovery of new reaction pathways, or the design of new molecules. The machine needs to read and write fluently in a chemical language for each of these tasks. Strings are a common tool to represent molecular graphs, and the most popular molecular string representation, SMILES, has powered cheminformatics since the late 1980s. However, in the context of AI and ML in chemistry, SMILES has several shortcomings -- most pertinently, most combinations of symbols lead to invalid results with no valid chemical interpretation. To overcome this issue, a new language for molecules was introduced in 2020 that guarantees 100\% robustness: SELFIES (SELF-referencIng Embedded Strings). SELFIES has since simplified and enabled numerous new applications in chemistry. In this manuscript, we look to the future and discuss molecular string representations, along with their respective opportunities and challenges. We propose 16 concrete Future Projects for robust molecular representations. These involve the extension toward new chemical domains, exciting questions at the interface of AI and robust languages and interpretability for both humans and machines. We hope that these proposals will inspire several follow-up works exploiting the full potential of molecular string representations for the future of AI in chemistry and materials science.