 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeConcept Bottleneck Language Models For protein design
Nov 09, 2024



We introduce Concept Bottleneck Protein Language Models (CB-pLM), a generative masked language model with a layer where each neuron corresponds to an interpretable concept. Our architecture offers three key benefits: i) Control: We can intervene on concept values to precisely control the properties of generated proteins, achieving a 3 times larger change in desired concept values compared to baselines. ii) Interpretability: A linear mapping between concept values and predicted tokens allows transparent analysis of the model's decision-making process. iii) Debugging: This transparency facilitates easy debugging of trained models. Our models achieve pre-training perplexity and downstream task performance comparable to traditional masked protein language models, demonstrating that interpretability does not compromise performance. While adaptable to any language model, we focus on masked protein language models due to their importance in drug discovery and the ability to validate our model's capabilities through real-world experiments and expert knowledge. We scale our CB-pLM from 24 million to 3 billion parameters, making them the largest Concept Bottleneck Models trained and the first capable of generative language modeling.
JAMUN: Transferable Molecular Conformational Ensemble Generation with Walk-Jump Sampling
Oct 18, 2024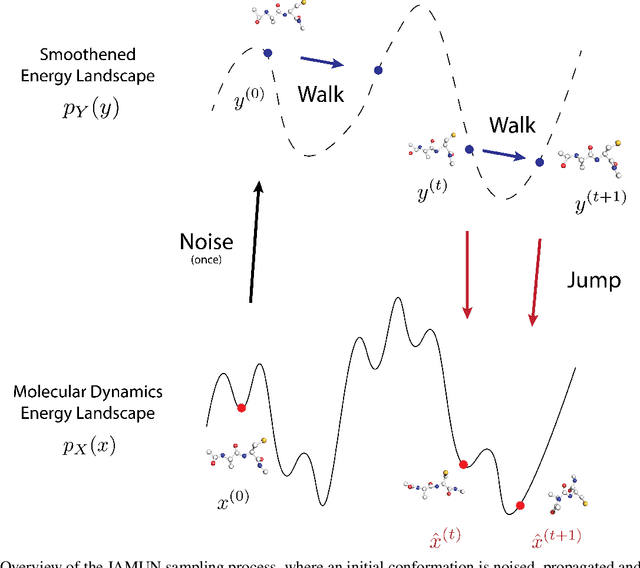

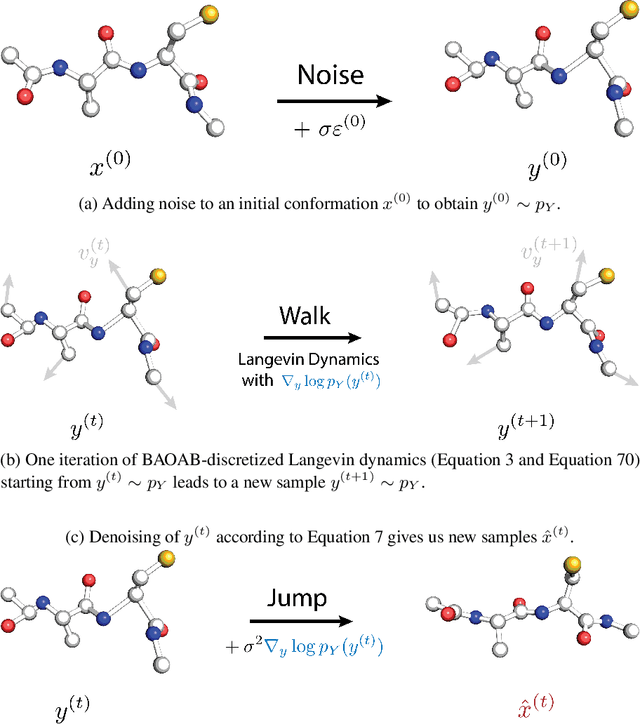
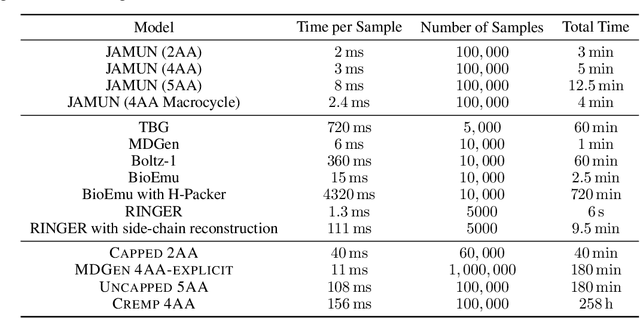
Conformational ensembles of protein structures are immensely important both to understanding protein function, and for drug discovery in novel modalities such as cryptic pockets. Current techniques for sampling ensembles are computationally inefficient, or do not transfer to systems outside their training data. We present walk-Jump Accelerated Molecular ensembles with Universal Noise (JAMUN), a step towards the goal of efficiently sampling the Boltzmann distribution of arbitrary proteins. By extending Walk-Jump Sampling to point clouds, JAMUN enables ensemble generation at orders of magnitude faster rates than traditional molecular dynamics or state-of-the-art ML methods. Further, JAMUN is able to predict the stable basins of small peptides that were not seen during training.
3D molecule generation by denoising voxel grids
Jun 13, 2023We propose a new score-based approach to generate 3D molecules represented as atomic densities on regular grids. First, we train a denoising neural network that learns to map from a smooth distribution of noisy molecules to the distribution of real molecules. Then, we follow the neural empirical Bayes framework [Saremi and Hyvarinen, 2019] and generate molecules in two steps: (i) sample noisy density grids from a smooth distribution via underdamped Langevin Markov chain Monte Carlo, and (ii) recover the ``clean'' molecule by denoising the noisy grid with a single step. Our method, VoxMol, generates molecules in a fundamentally different way than the current state of the art (i.e., diffusion models applied to atom point clouds). It differs in terms of the data representation, the noise model, the network architecture and the generative modeling algorithm. VoxMol achieves comparable results to state of the art on unconditional 3D molecule generation while being simpler to train and faster to generate molecules.
Protein Discovery with Discrete Walk-Jump Sampling
Jun 08, 2023



We resolve difficulties in training and sampling from a discrete generative model by learning a smoothed energy function, sampling from the smoothed data manifold with Langevin Markov chain Monte Carlo (MCMC), and projecting back to the true data manifold with one-step denoising. Our Discrete Walk-Jump Sampling formalism combines the maximum likelihood training of an energy-based model and improved sample quality of a score-based model, while simplifying training and sampling by requiring only a single noise level. We evaluate the robustness of our approach on generative modeling of antibody proteins and introduce the distributional conformity score to benchmark protein generative models. By optimizing and sampling from our models for the proposed distributional conformity score, 97-100% of generated samples are successfully expressed and purified and 35% of functional designs show equal or improved binding affinity compared to known functional antibodies on the first attempt in a single round of laboratory experiments. We also report the first demonstration of long-run fast-mixing MCMC chains where diverse antibody protein classes are visited in a single MCMC chain.