 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLLMs are Highly-Constrained Biophysical Sequence Optimizers
Oct 29, 2024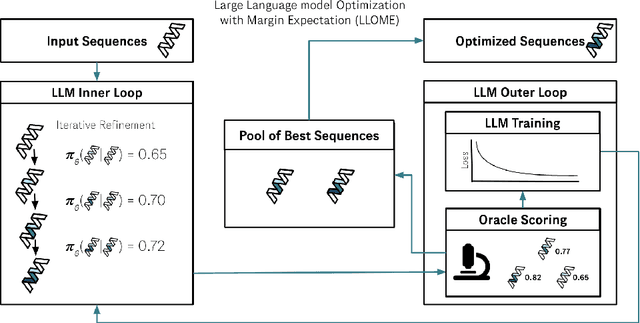
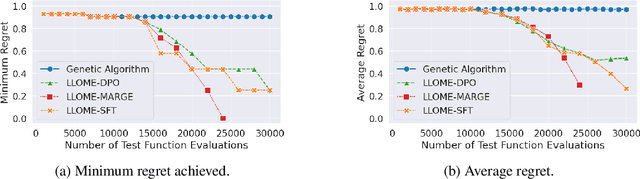
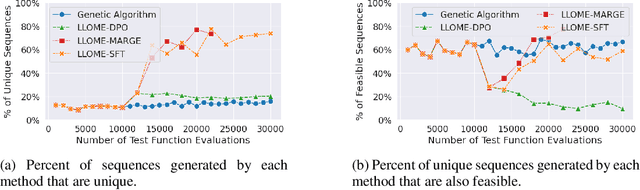
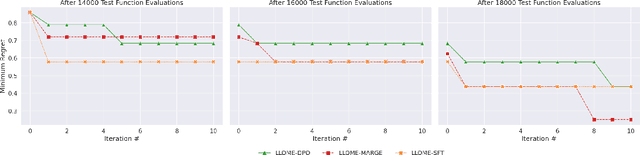
Large language models (LLMs) have recently shown significant potential in various biological tasks such as protein engineering and molecule design. These tasks typically involve black-box discrete sequence optimization, where the challenge lies in generating sequences that are not only biologically feasible but also adhere to hard fine-grained constraints. However, LLMs often struggle with such constraints, especially in biological contexts where verifying candidate solutions is costly and time-consuming. In this study, we explore the possibility of employing LLMs as highly-constrained bilevel optimizers through a methodology we refer to as Language Model Optimization with Margin Expectation (LLOME). This approach combines both offline and online optimization, utilizing limited oracle evaluations to iteratively enhance the sequences generated by the LLM. We additionally propose a novel training objective -- Margin-Aligned Expectation (MargE) -- that trains the LLM to smoothly interpolate between the reward and reference distributions. Lastly, we introduce a synthetic test suite that bears strong geometric similarity to real biophysical problems and enables rapid evaluation of LLM optimizers without time-consuming lab validation. Our findings reveal that, in comparison to genetic algorithm baselines, LLMs achieve significantly lower regret solutions while requiring fewer test function evaluations. However, we also observe that LLMs exhibit moderate miscalibration, are susceptible to generator collapse, and have difficulty finding the optimal solution when no explicit ground truth rewards are available.
OpenProteinSet: Training data for structural biology at scale
Aug 10, 2023


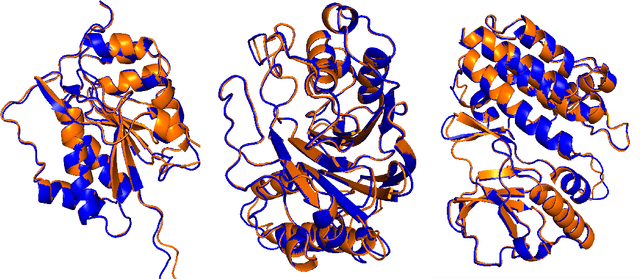
Multiple sequence alignments (MSAs) of proteins encode rich biological information and have been workhorses in bioinformatic methods for tasks like protein design and protein structure prediction for decades. Recent breakthroughs like AlphaFold2 that use transformers to attend directly over large quantities of raw MSAs have reaffirmed their importance. Generation of MSAs is highly computationally intensive, however, and no datasets comparable to those used to train AlphaFold2 have been made available to the research community, hindering progress in machine learning for proteins. To remedy this problem, we introduce OpenProteinSet, an open-source corpus of more than 16 million MSAs, associated structural homologs from the Protein Data Bank, and AlphaFold2 protein structure predictions. We have previously demonstrated the utility of OpenProteinSet by successfully retraining AlphaFold2 on it. We expect OpenProteinSet to be broadly useful as training and validation data for 1) diverse tasks focused on protein structure, function, and design and 2) large-scale multimodal machine learning research.
AbDiffuser: Full-Atom Generation of In-Vitro Functioning Antibodies
Jul 28, 2023



We introduce AbDiffuser, an equivariant and physics-informed diffusion model for the joint generation of antibody 3D structures and sequences. AbDiffuser is built on top of a new representation of protein structure, relies on a novel architecture for aligned proteins, and utilizes strong diffusion priors to improve the denoising process. Our approach improves protein diffusion by taking advantage of domain knowledge and physics-based constraints; handles sequence-length changes; and reduces memory complexity by an order of magnitude enabling backbone and side chain generation. We validate AbDiffuser in silico and in vitro. Numerical experiments showcase the ability of AbDiffuser to generate antibodies that closely track the sequence and structural properties of a reference set. Laboratory experiments confirm that all 16 HER2 antibodies discovered were expressed at high levels and that 57.1% of selected designs were tight binders.
Protein Discovery with Discrete Walk-Jump Sampling
Jun 08, 2023



We resolve difficulties in training and sampling from a discrete generative model by learning a smoothed energy function, sampling from the smoothed data manifold with Langevin Markov chain Monte Carlo (MCMC), and projecting back to the true data manifold with one-step denoising. Our Discrete Walk-Jump Sampling formalism combines the maximum likelihood training of an energy-based model and improved sample quality of a score-based model, while simplifying training and sampling by requiring only a single noise level. We evaluate the robustness of our approach on generative modeling of antibody proteins and introduce the distributional conformity score to benchmark protein generative models. By optimizing and sampling from our models for the proposed distributional conformity score, 97-100% of generated samples are successfully expressed and purified and 35% of functional designs show equal or improved binding affinity compared to known functional antibodies on the first attempt in a single round of laboratory experiments. We also report the first demonstration of long-run fast-mixing MCMC chains where diverse antibody protein classes are visited in a single MCMC chain.
Dictionary-Assisted Supervised Contrastive Learning
Oct 27, 2022
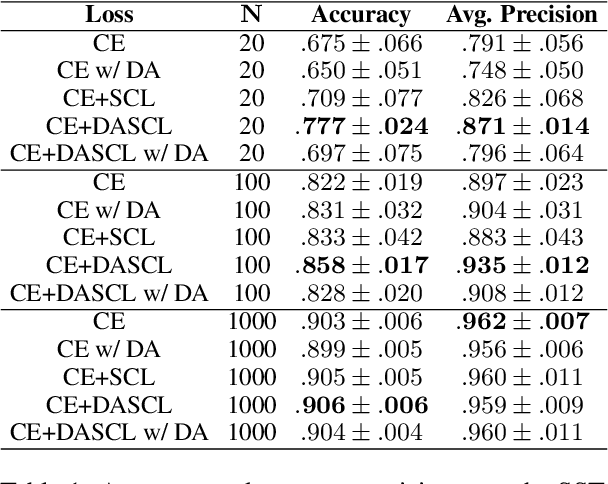
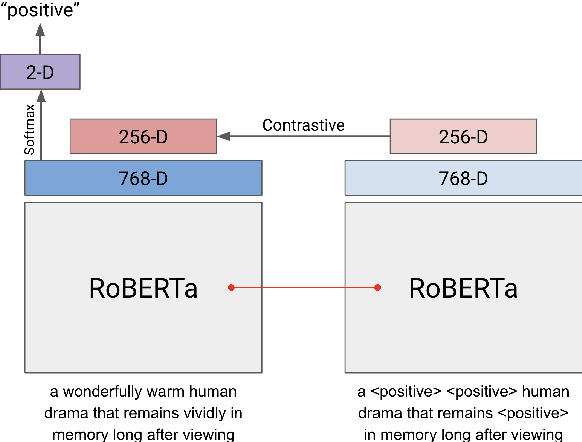
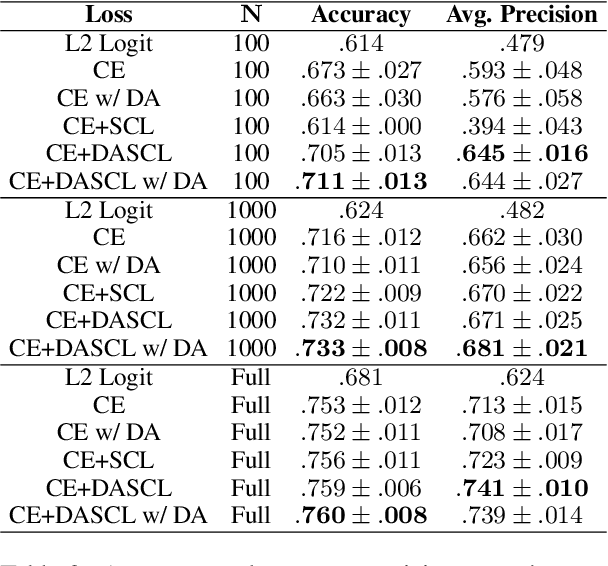
Text analysis in the social sciences often involves using specialized dictionaries to reason with abstract concepts, such as perceptions about the economy or abuse on social media. These dictionaries allow researchers to impart domain knowledge and note subtle usages of words relating to a concept(s) of interest. We introduce the dictionary-assisted supervised contrastive learning (DASCL) objective, allowing researchers to leverage specialized dictionaries when fine-tuning pretrained language models. The text is first keyword simplified: a common, fixed token replaces any word in the corpus that appears in the dictionary(ies) relevant to the concept of interest. During fine-tuning, a supervised contrastive objective draws closer the embeddings of the original and keyword-simplified texts of the same class while pushing further apart the embeddings of different classes. The keyword-simplified texts of the same class are more textually similar than their original text counterparts, which additionally draws the embeddings of the same class closer together. Combining DASCL and cross-entropy improves classification performance metrics in few-shot learning settings and social science applications compared to using cross-entropy alone and alternative contrastive and data augmentation methods.
A Pareto-optimal compositional energy-based model for sampling and optimization of protein sequences
Oct 19, 2022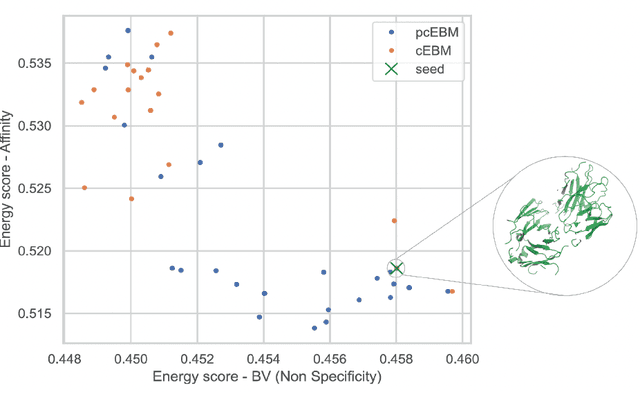
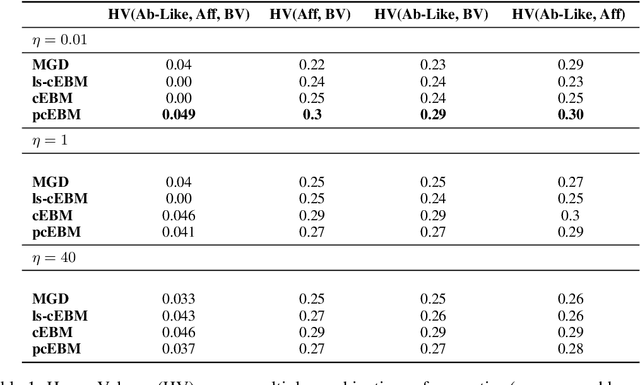
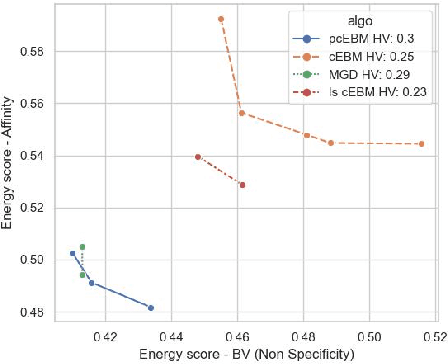
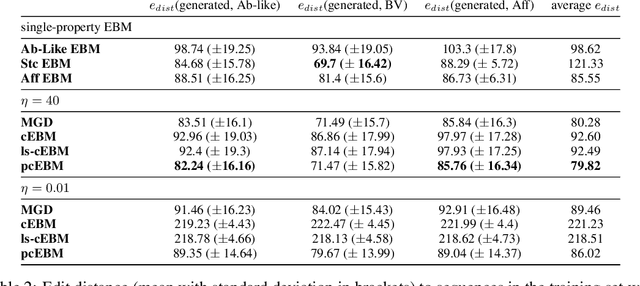
Deep generative models have emerged as a popular machine learning-based approach for inverse design problems in the life sciences. However, these problems often require sampling new designs that satisfy multiple properties of interest in addition to learning the data distribution. This multi-objective optimization becomes more challenging when properties are independent or orthogonal to each other. In this work, we propose a Pareto-compositional energy-based model (pcEBM), a framework that uses multiple gradient descent for sampling new designs that adhere to various constraints in optimizing distinct properties. We demonstrate its ability to learn non-convex Pareto fronts and generate sequences that simultaneously satisfy multiple desired properties across a series of real-world antibody design tasks.
PropertyDAG: Multi-objective Bayesian optimization of partially ordered, mixed-variable properties for biological sequence design
Oct 08, 2022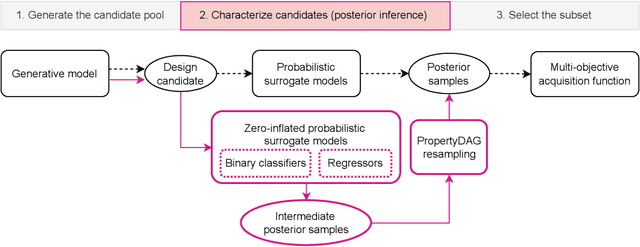
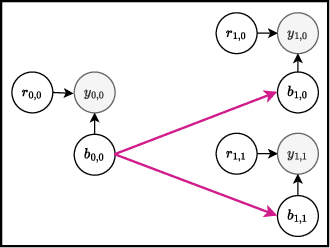
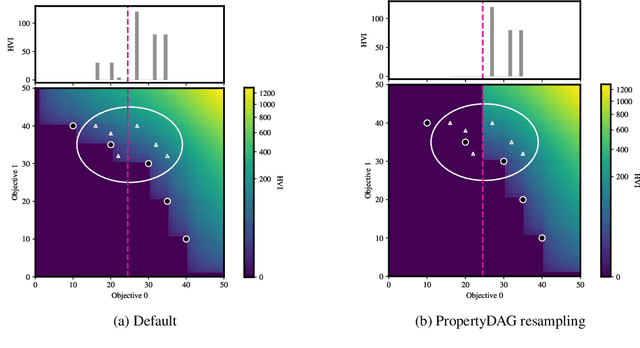
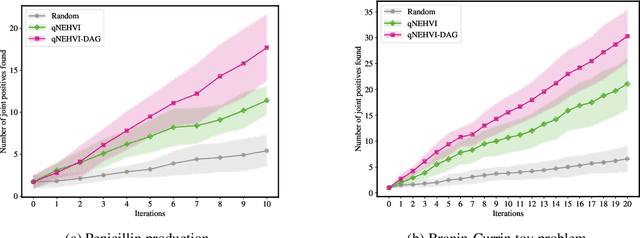
Bayesian optimization offers a sample-efficient framework for navigating the exploration-exploitation trade-off in the vast design space of biological sequences. Whereas it is possible to optimize the various properties of interest jointly using a multi-objective acquisition function, such as the expected hypervolume improvement (EHVI), this approach does not account for objectives with a hierarchical dependency structure. We consider a common use case where some regions of the Pareto frontier are prioritized over others according to a specified $\textit{partial ordering}$ in the objectives. For instance, when designing antibodies, we would like to maximize the binding affinity to a target antigen only if it can be expressed in live cell culture -- modeling the experimental dependency in which affinity can only be measured for antibodies that can be expressed and thus produced in viable quantities. In general, we may want to confer a partial ordering to the properties such that each property is optimized conditioned on its parent properties satisfying some feasibility condition. To this end, we present PropertyDAG, a framework that operates on top of the traditional multi-objective BO to impose this desired ordering on the objectives, e.g. expression $\rightarrow$ affinity. We demonstrate its performance over multiple simulated active learning iterations on a penicillin production task, toy numerical problem, and a real-world antibody design task.
Multi-segment preserving sampling for deep manifold sampler
May 09, 2022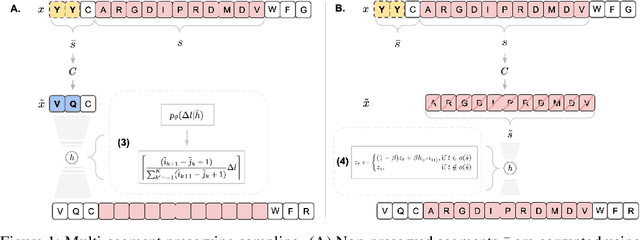

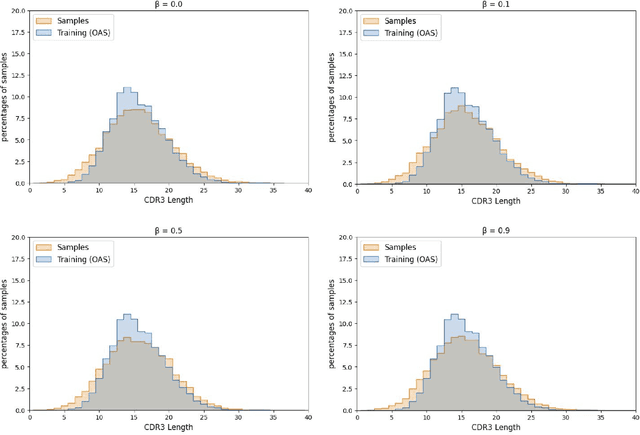
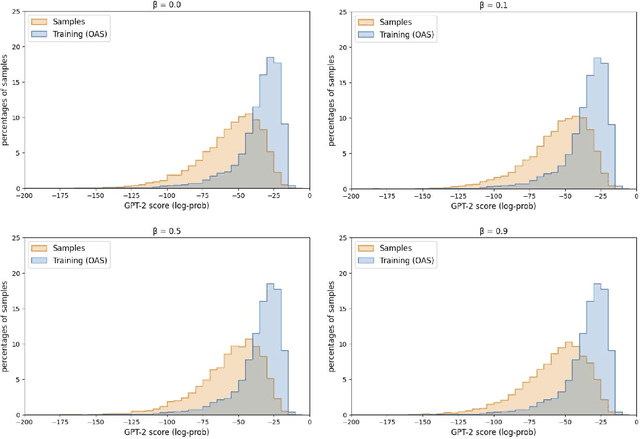
Deep generative modeling for biological sequences presents a unique challenge in reconciling the bias-variance trade-off between explicit biological insight and model flexibility. The deep manifold sampler was recently proposed as a means to iteratively sample variable-length protein sequences by exploiting the gradients from a function predictor. We introduce an alternative approach to this guided sampling procedure, multi-segment preserving sampling, that enables the direct inclusion of domain-specific knowledge by designating preserved and non-preserved segments along the input sequence, thereby restricting variation to only select regions. We present its effectiveness in the context of antibody design by training two models: a deep manifold sampler and a GPT-2 language model on nearly six million heavy chain sequences annotated with the IGHV1-18 gene. During sampling, we restrict variation to only the complementarity-determining region 3 (CDR3) of the input. We obtain log probability scores from a GPT-2 model for each sampled CDR3 and demonstrate that multi-segment preserving sampling generates reasonable designs while maintaining the desired, preserved regions.