 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBlind Biological Sequence Denoising with Self-Supervised Set Learning
Sep 04, 2023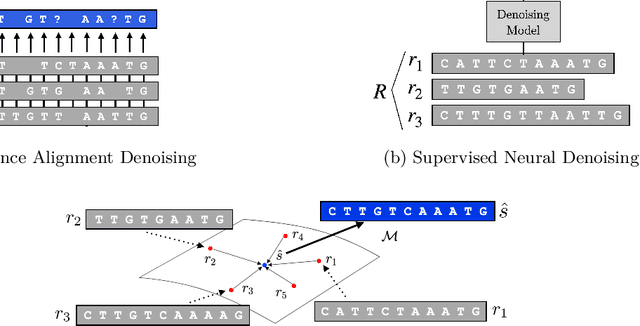

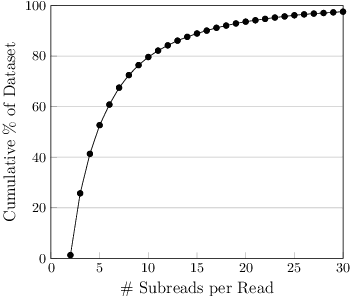

Biological sequence analysis relies on the ability to denoise the imprecise output of sequencing platforms. We consider a common setting where a short sequence is read out repeatedly using a high-throughput long-read platform to generate multiple subreads, or noisy observations of the same sequence. Denoising these subreads with alignment-based approaches often fails when too few subreads are available or error rates are too high. In this paper, we propose a novel method for blindly denoising sets of sequences without directly observing clean source sequence labels. Our method, Self-Supervised Set Learning (SSSL), gathers subreads together in an embedding space and estimates a single set embedding as the midpoint of the subreads in both the latent and sequence spaces. This set embedding represents the "average" of the subreads and can be decoded into a prediction of the clean sequence. In experiments on simulated long-read DNA data, SSSL methods denoise small reads of $\leq 6$ subreads with 17% fewer errors and large reads of $>6$ subreads with 8% fewer errors compared to the best baseline. On a real dataset of antibody sequences, SSSL improves over baselines on two self-supervised metrics, with a significant improvement on difficult small reads that comprise over 60% of the test set. By accurately denoising these reads, SSSL promises to better realize the potential of high-throughput DNA sequencing data for downstream scientific applications.
MoleCLUEs: Optimizing Molecular Conformers by Minimization of Differentiable Uncertainty
Jun 20, 2023



Structure-based models in the molecular sciences can be highly sensitive to input geometries and give predictions with large variance under subtle coordinate perturbations. We present an approach to mitigate this failure mode by generating conformations that explicitly minimize uncertainty in a predictive model. To achieve this, we compute differentiable estimates of aleatoric \textit{and} epistemic uncertainties directly from learned embeddings. We then train an optimizer that iteratively samples embeddings to reduce these uncertainties according to their gradients. As our predictive model is constructed as a variational autoencoder, the new embeddings can be decoded to their corresponding inputs, which we call \textit{MoleCLUEs}, or (molecular) counterfactual latent uncertainty explanations \citep{antoran2020getting}. We provide results of our algorithm for the task of predicting drug properties with maximum confidence as well as analysis of the differentiable structure simulations.
SupSiam: Non-contrastive Auxiliary Loss for Learning from Molecular Conformers
Feb 15, 2023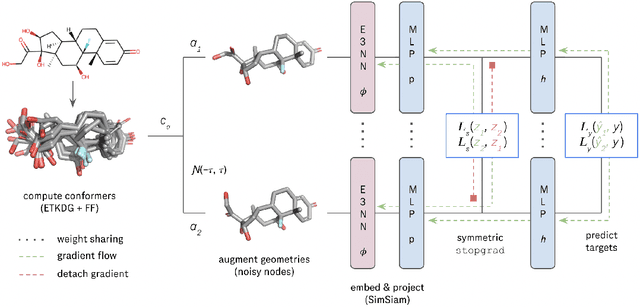

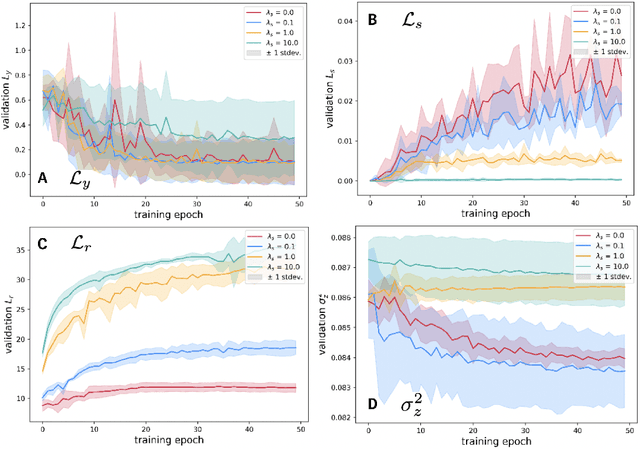

We investigate Siamese networks for learning related embeddings for augmented samples of molecular conformers. We find that a non-contrastive (positive-pair only) auxiliary task aids in supervised training of Euclidean neural networks (E3NNs) and increases manifold smoothness (MS) around point-cloud geometries. We demonstrate this property for multiple drug-activity prediction tasks while maintaining relevant performance metrics, and propose an extension of MS to probabilistic and regression settings. We provide an analysis of representation collapse, finding substantial effects of task-weighting, latent dimension, and regularization. We expect the presented protocol to aid in the development of reliable E3NNs from molecular conformers, even for small-data drug discovery programs.
Multi-segment preserving sampling for deep manifold sampler
May 09, 2022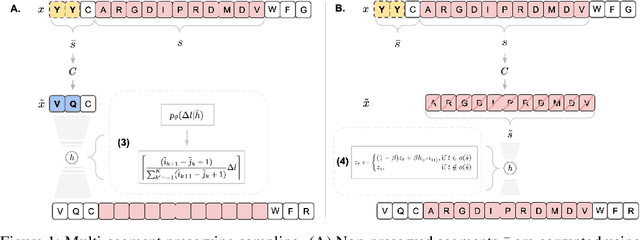

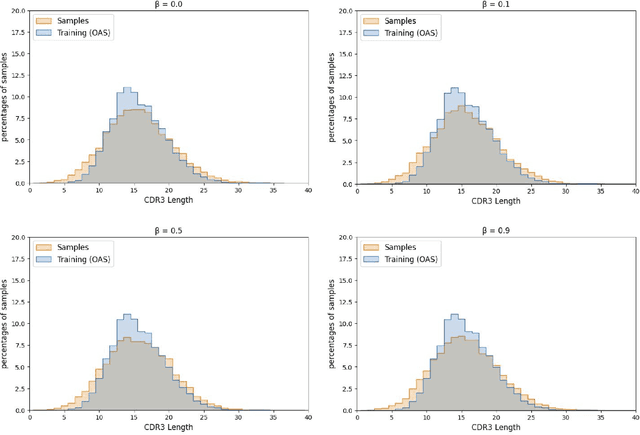
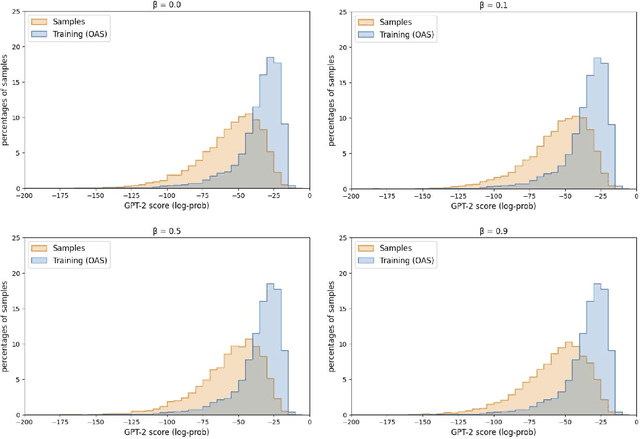
Deep generative modeling for biological sequences presents a unique challenge in reconciling the bias-variance trade-off between explicit biological insight and model flexibility. The deep manifold sampler was recently proposed as a means to iteratively sample variable-length protein sequences by exploiting the gradients from a function predictor. We introduce an alternative approach to this guided sampling procedure, multi-segment preserving sampling, that enables the direct inclusion of domain-specific knowledge by designating preserved and non-preserved segments along the input sequence, thereby restricting variation to only select regions. We present its effectiveness in the context of antibody design by training two models: a deep manifold sampler and a GPT-2 language model on nearly six million heavy chain sequences annotated with the IGHV1-18 gene. During sampling, we restrict variation to only the complementarity-determining region 3 (CDR3) of the input. We obtain log probability scores from a GPT-2 model for each sampled CDR3 and demonstrate that multi-segment preserving sampling generates reasonable designs while maintaining the desired, preserved regions.