 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOpenProteinSet: Training data for structural biology at scale
Aug 10, 2023


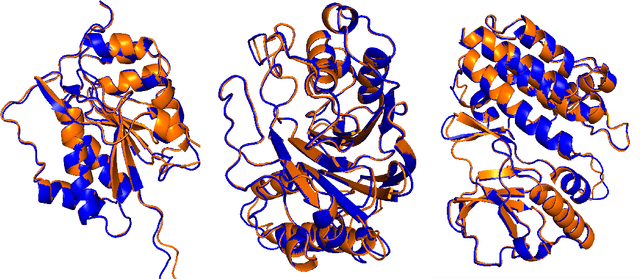
Multiple sequence alignments (MSAs) of proteins encode rich biological information and have been workhorses in bioinformatic methods for tasks like protein design and protein structure prediction for decades. Recent breakthroughs like AlphaFold2 that use transformers to attend directly over large quantities of raw MSAs have reaffirmed their importance. Generation of MSAs is highly computationally intensive, however, and no datasets comparable to those used to train AlphaFold2 have been made available to the research community, hindering progress in machine learning for proteins. To remedy this problem, we introduce OpenProteinSet, an open-source corpus of more than 16 million MSAs, associated structural homologs from the Protein Data Bank, and AlphaFold2 protein structure predictions. We have previously demonstrated the utility of OpenProteinSet by successfully retraining AlphaFold2 on it. We expect OpenProteinSet to be broadly useful as training and validation data for 1) diverse tasks focused on protein structure, function, and design and 2) large-scale multimodal machine learning research.
Protein Discovery with Discrete Walk-Jump Sampling
Jun 08, 2023



We resolve difficulties in training and sampling from a discrete generative model by learning a smoothed energy function, sampling from the smoothed data manifold with Langevin Markov chain Monte Carlo (MCMC), and projecting back to the true data manifold with one-step denoising. Our Discrete Walk-Jump Sampling formalism combines the maximum likelihood training of an energy-based model and improved sample quality of a score-based model, while simplifying training and sampling by requiring only a single noise level. We evaluate the robustness of our approach on generative modeling of antibody proteins and introduce the distributional conformity score to benchmark protein generative models. By optimizing and sampling from our models for the proposed distributional conformity score, 97-100% of generated samples are successfully expressed and purified and 35% of functional designs show equal or improved binding affinity compared to known functional antibodies on the first attempt in a single round of laboratory experiments. We also report the first demonstration of long-run fast-mixing MCMC chains where diverse antibody protein classes are visited in a single MCMC chain.
Multi-segment preserving sampling for deep manifold sampler
May 09, 2022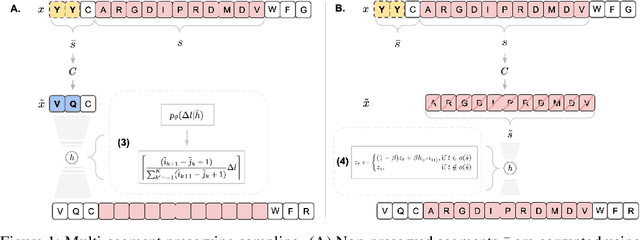

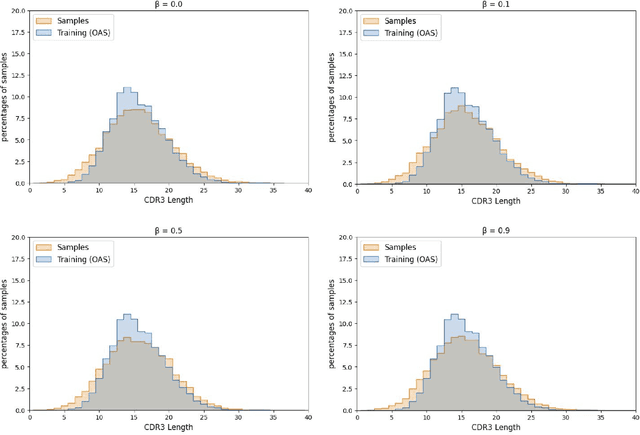
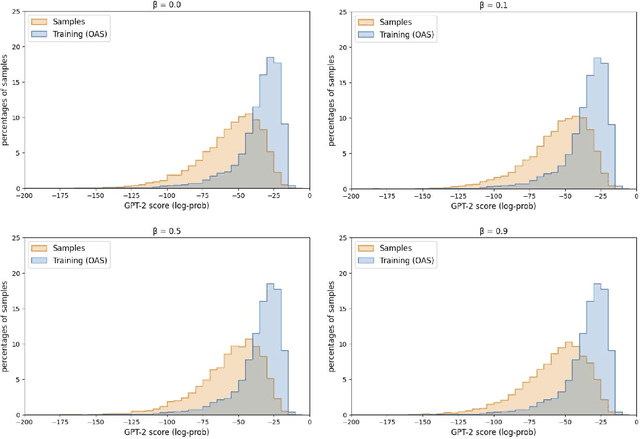
Deep generative modeling for biological sequences presents a unique challenge in reconciling the bias-variance trade-off between explicit biological insight and model flexibility. The deep manifold sampler was recently proposed as a means to iteratively sample variable-length protein sequences by exploiting the gradients from a function predictor. We introduce an alternative approach to this guided sampling procedure, multi-segment preserving sampling, that enables the direct inclusion of domain-specific knowledge by designating preserved and non-preserved segments along the input sequence, thereby restricting variation to only select regions. We present its effectiveness in the context of antibody design by training two models: a deep manifold sampler and a GPT-2 language model on nearly six million heavy chain sequences annotated with the IGHV1-18 gene. During sampling, we restrict variation to only the complementarity-determining region 3 (CDR3) of the input. We obtain log probability scores from a GPT-2 model for each sampled CDR3 and demonstrate that multi-segment preserving sampling generates reasonable designs while maintaining the desired, preserved regions.
Inferring the size of the causal universe: features and fusion of causal attribution networks
Dec 14, 2018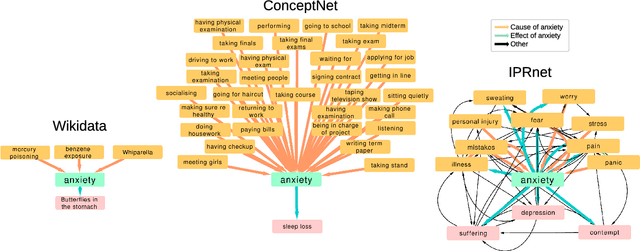
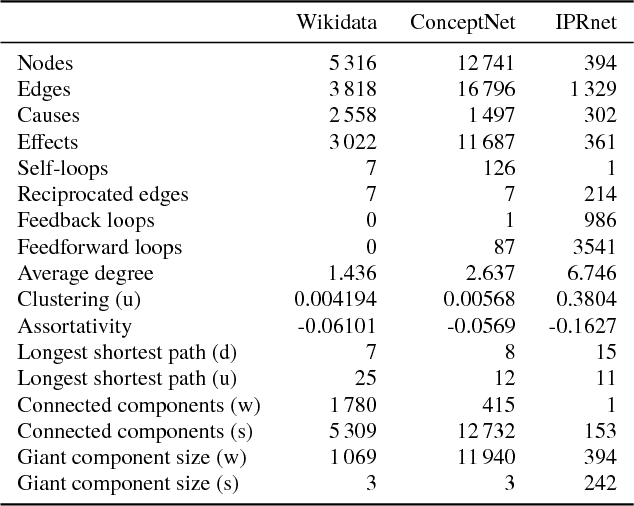
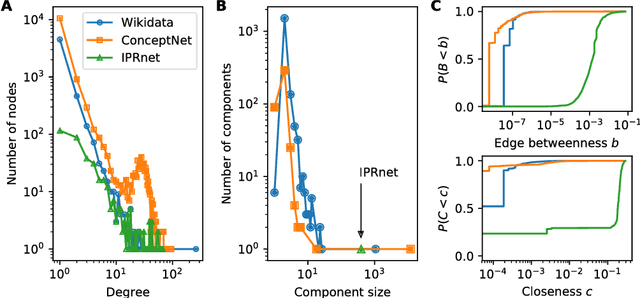
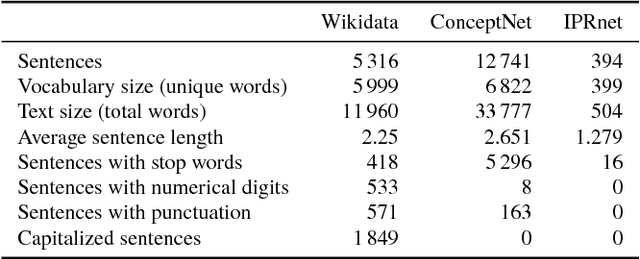
Cause-and-effect reasoning, the attribution of effects to causes, is one of the most powerful and unique skills humans possess. Multiple surveys are mapping out causal attributions as networks, but it is unclear how well these efforts can be combined. Further, the total size of the collective causal attribution network held by humans is currently unknown, making it challenging to assess the progress of these surveys. Here we study three causal attribution networks to determine how well they can be combined into a single network. Combining these networks requires dealing with ambiguous nodes, as nodes represent written descriptions of causes and effects and different descriptions may exist for the same concept. We introduce NetFUSES, a method for combining networks with ambiguous nodes. Crucially, treating the different causal attributions networks as independent samples allows us to use their overlap to estimate the total size of the collective causal attribution network. We find that existing surveys capture 5.77% $\pm$ 0.781% of the $\approx$293 000 causes and effects estimated to exist, and 0.198% $\pm$ 0.174% of the $\approx$10 200 000 attributed cause-effect relationships.
Neural language representations predict outcomes of scientific research
May 17, 2018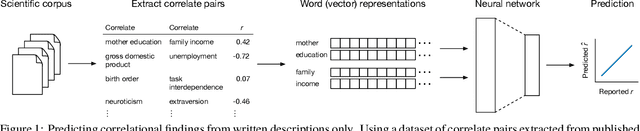
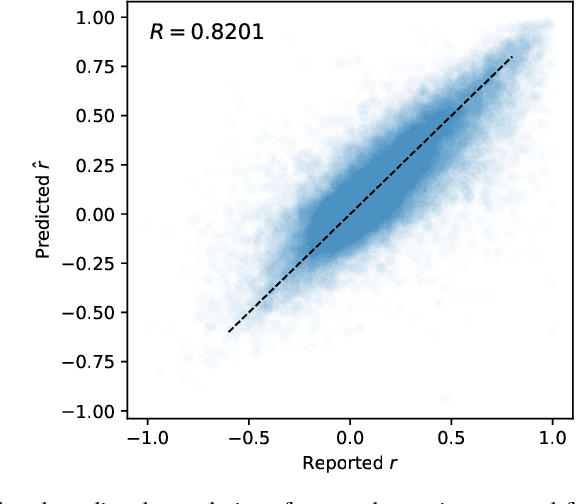
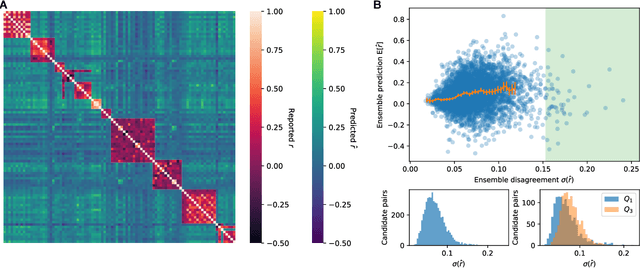
Many research fields codify their findings in standard formats, often by reporting correlations between quantities of interest. But the space of all testable correlates is far larger than scientific resources can currently address, so the ability to accurately predict correlations would be useful to plan research and allocate resources. Using a dataset of approximately 170,000 correlational findings extracted from leading social science journals, we show that a trained neural network can accurately predict the reported correlations using only the text descriptions of the correlates. Accurate predictive models such as these can guide scientists towards promising untested correlates, better quantify the information gained from new findings, and has implications for moving artificial intelligence systems from predicting structures to predicting relationships in the real world.