 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeQuickBind: A Light-Weight And Interpretable Molecular Docking Model
Oct 21, 2024
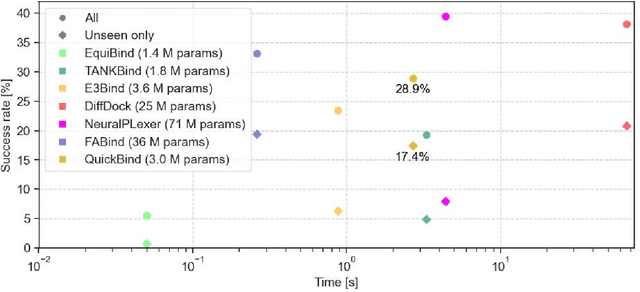
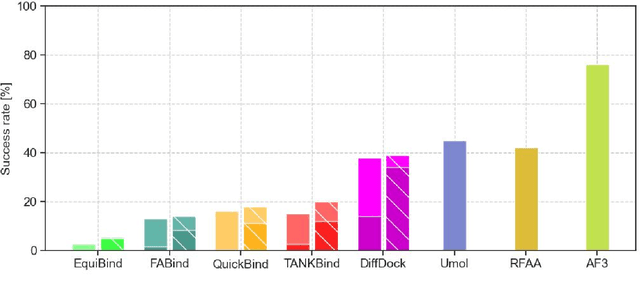

Predicting a ligand's bound pose to a target protein is a key component of early-stage computational drug discovery. Recent developments in machine learning methods have focused on improving pose quality at the cost of model runtime. For high-throughput virtual screening applications, this exposes a capability gap that can be filled by moderately accurate but fast pose prediction. To this end, we developed QuickBind, a light-weight pose prediction algorithm. We assess QuickBind on widely used benchmarks and find that it provides an attractive trade-off between model accuracy and runtime. To facilitate virtual screening applications, we augment QuickBind with a binding affinity module and demonstrate its capabilities for multiple clinically-relevant drug targets. Finally, we investigate the mechanistic basis by which QuickBind makes predictions and find that it has learned key physicochemical properties of molecular docking, providing new insights into how machine learning models generate protein-ligand poses. By virtue of its simplicity, QuickBind can serve as both an effective virtual screening tool and a minimal test bed for exploring new model architectures and innovations. Model code and weights are available at https://github.com/aqlaboratory/QuickBind .
How to Build the Virtual Cell with Artificial Intelligence: Priorities and Opportunities
Sep 18, 2024

The cell is arguably the smallest unit of life and is central to understanding biology. Accurate modeling of cells is important for this understanding as well as for determining the root causes of disease. Recent advances in artificial intelligence (AI), combined with the ability to generate large-scale experimental data, present novel opportunities to model cells. Here we propose a vision of AI-powered Virtual Cells, where robust representations of cells and cellular systems under different conditions are directly learned from growing biological data across measurements and scales. We discuss desired capabilities of AI Virtual Cells, including generating universal representations of biological entities across scales, and facilitating interpretable in silico experiments to predict and understand their behavior using Virtual Instruments. We further address the challenges, opportunities and requirements to realize this vision including data needs, evaluation strategies, and community standards and engagement to ensure biological accuracy and broad utility. We envision a future where AI Virtual Cells help identify new drug targets, predict cellular responses to perturbations, as well as scale hypothesis exploration. With open science collaborations across the biomedical ecosystem that includes academia, philanthropy, and the biopharma and AI industries, a comprehensive predictive understanding of cell mechanisms and interactions is within reach.
Out of Many, One: Designing and Scaffolding Proteins at the Scale of the Structural Universe with Genie 2
May 24, 2024



Protein diffusion models have emerged as a promising approach for protein design. One such pioneering model is Genie, a method that asymmetrically represents protein structures during the forward and backward processes, using simple Gaussian noising for the former and expressive SE(3)-equivariant attention for the latter. In this work we introduce Genie 2, extending Genie to capture a larger and more diverse protein structure space through architectural innovations and massive data augmentation. Genie 2 adds motif scaffolding capabilities via a novel multi-motif framework that designs co-occurring motifs with unspecified inter-motif positions and orientations. This makes possible complex protein designs that engage multiple interaction partners and perform multiple functions. On both unconditional and conditional generation, Genie 2 achieves state-of-the-art performance, outperforming all known methods on key design metrics including designability, diversity, and novelty. Genie 2 also solves more motif scaffolding problems than other methods and does so with more unique and varied solutions. Taken together, these advances set a new standard for structure-based protein design. Genie 2 inference and training code, as well as model weights, are freely available at: https://github.com/aqlaboratory/genie2.
DeepSpeed4Science Initiative: Enabling Large-Scale Scientific Discovery through Sophisticated AI System Technologies
Oct 11, 2023



In the upcoming decade, deep learning may revolutionize the natural sciences, enhancing our capacity to model and predict natural occurrences. This could herald a new era of scientific exploration, bringing significant advancements across sectors from drug development to renewable energy. To answer this call, we present DeepSpeed4Science initiative (deepspeed4science.ai) which aims to build unique capabilities through AI system technology innovations to help domain experts to unlock today's biggest science mysteries. By leveraging DeepSpeed's current technology pillars (training, inference and compression) as base technology enablers, DeepSpeed4Science will create a new set of AI system technologies tailored for accelerating scientific discoveries by addressing their unique complexity beyond the common technical approaches used for accelerating generic large language models (LLMs). In this paper, we showcase the early progress we made with DeepSpeed4Science in addressing two of the critical system challenges in structural biology research.
Growing ecosystem of deep learning methods for modeling protein$\unicode{x2013}$protein interactions
Oct 10, 2023



Numerous cellular functions rely on protein$\unicode{x2013}$protein interactions. Efforts to comprehensively characterize them remain challenged however by the diversity of molecular recognition mechanisms employed within the proteome. Deep learning has emerged as a promising approach for tackling this problem by exploiting both experimental data and basic biophysical knowledge about protein interactions. Here, we review the growing ecosystem of deep learning methods for modeling protein interactions, highlighting the diversity of these biophysically-informed models and their respective trade-offs. We discuss recent successes in using representation learning to capture complex features pertinent to predicting protein interactions and interaction sites, geometric deep learning to reason over protein structures and predict complex structures, and generative modeling to design de novo protein assemblies. We also outline some of the outstanding challenges and promising new directions. Opportunities abound to discover novel interactions, elucidate their physical mechanisms, and engineer binders to modulate their functions using deep learning and, ultimately, unravel how protein interactions orchestrate complex cellular behaviors.
OpenProteinSet: Training data for structural biology at scale
Aug 10, 2023


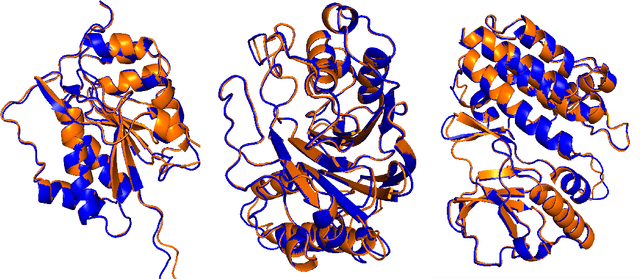
Multiple sequence alignments (MSAs) of proteins encode rich biological information and have been workhorses in bioinformatic methods for tasks like protein design and protein structure prediction for decades. Recent breakthroughs like AlphaFold2 that use transformers to attend directly over large quantities of raw MSAs have reaffirmed their importance. Generation of MSAs is highly computationally intensive, however, and no datasets comparable to those used to train AlphaFold2 have been made available to the research community, hindering progress in machine learning for proteins. To remedy this problem, we introduce OpenProteinSet, an open-source corpus of more than 16 million MSAs, associated structural homologs from the Protein Data Bank, and AlphaFold2 protein structure predictions. We have previously demonstrated the utility of OpenProteinSet by successfully retraining AlphaFold2 on it. We expect OpenProteinSet to be broadly useful as training and validation data for 1) diverse tasks focused on protein structure, function, and design and 2) large-scale multimodal machine learning research.
Generating Novel, Designable, and Diverse Protein Structures by Equivariantly Diffusing Oriented Residue Clouds
Feb 03, 2023Proteins power a vast array of functional processes in living cells. The capability to create new proteins with designed structures and functions would thus enable the engineering of cellular behavior and development of protein-based therapeutics and materials. Structure-based protein design aims to find structures that are designable (can be realized by a protein sequence), novel (have dissimilar geometry from natural proteins), and diverse (span a wide range of geometries). While advances in protein structure prediction have made it possible to predict structures of novel protein sequences, the combinatorially large space of sequences and structures limits the practicality of search-based methods. Generative models provide a compelling alternative, by implicitly learning the low-dimensional structure of complex data distributions. Here, we leverage recent advances in denoising diffusion probabilistic models and equivariant neural networks to develop Genie, a generative model of protein structures that performs discrete-time diffusion using a cloud of oriented reference frames in 3D space. Through in silico evaluations, we demonstrate that Genie generates protein backbones that are more designable, novel, and diverse than existing models. This indicates that Genie is capturing key aspects of the distribution of protein structure space and facilitates protein design with high success rates. Code for generating new proteins and training new versions of Genie is available at https://github.com/aqlaboratory/genie.
Accurate Protein Structure Prediction by Embeddings and Deep Learning Representations
Nov 09, 2019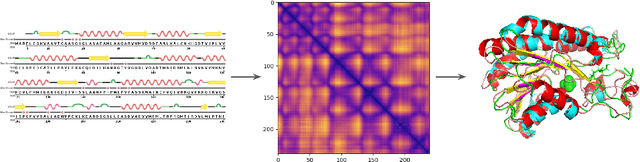


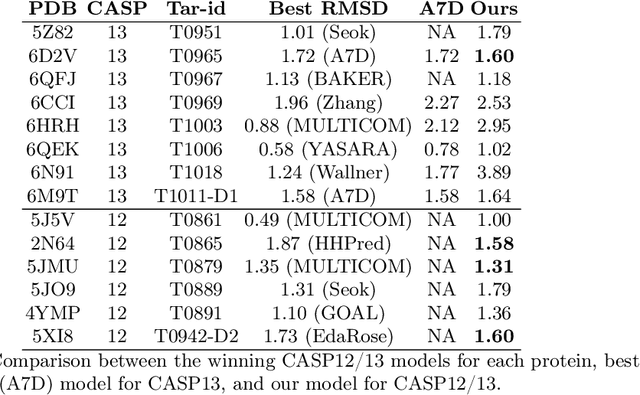
Proteins are the major building blocks of life, and actuators of almost all chemical and biophysical events in living organisms. Their native structures in turn enable their biological functions which have a fundamental role in drug design. This motivates predicting the structure of a protein from its sequence of amino acids, a fundamental problem in computational biology. In this work, we demonstrate state-of-the-art protein structure prediction (PSP) results using embeddings and deep learning models for prediction of backbone atom distance matrices and torsion angles. We recover 3D coordinates of backbone atoms and reconstruct full atom protein by optimization. We create a new gold standard dataset of proteins which is comprehensive and easy to use. Our dataset consists of amino acid sequences, Q8 secondary structures, position specific scoring matrices, multiple sequence alignment co-evolutionary features, backbone atom distance matrices, torsion angles, and 3D coordinates. We evaluate the quality of our structure prediction by RMSD on the latest Critical Assessment of Techniques for Protein Structure Prediction (CASP) test data and demonstrate competitive results with the winning teams and AlphaFold in CASP13 and supersede the results of the winning teams in CASP12. We make our data, models, and code publicly available.
ProteinNet: a standardized data set for machine learning of protein structure
Feb 01, 2019


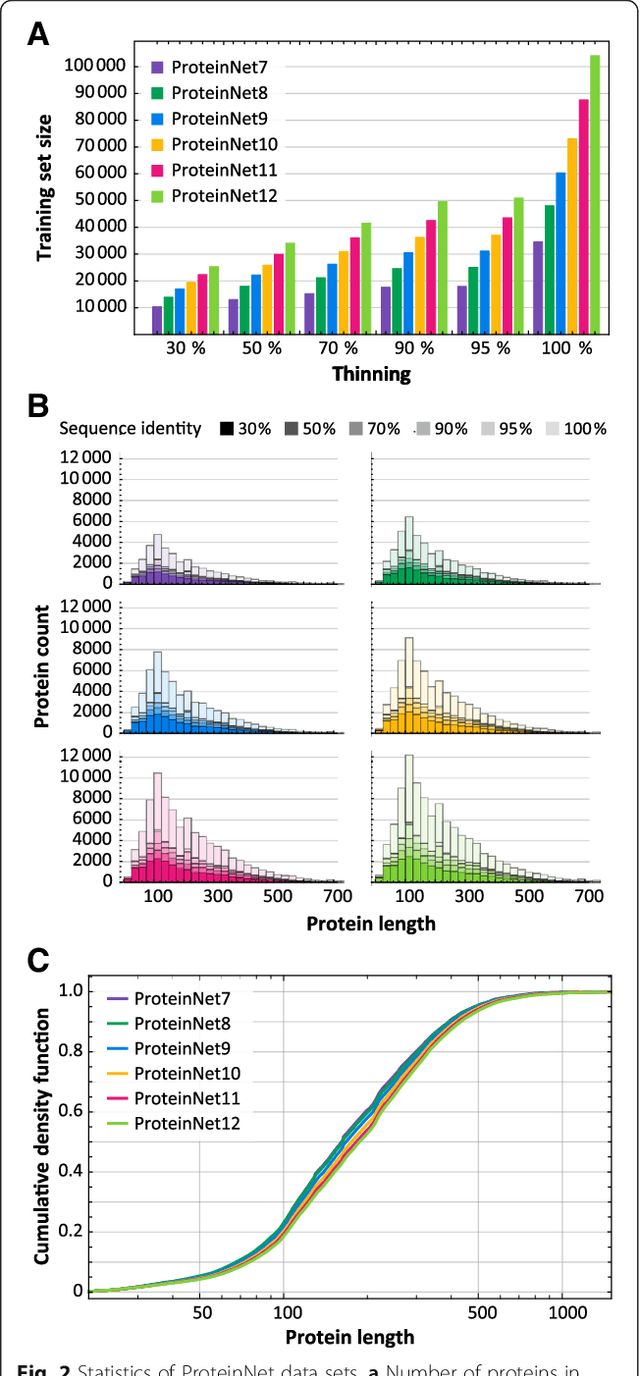
Rapid progress in deep learning has spurred its application to bioinformatics problems including protein structure prediction and design. In classic machine learning problems like computer vision, progress has been driven by standardized data sets that facilitate fair assessment of new methods and lower the barrier to entry for non-domain experts. While data sets of protein sequence and structure exist, they lack certain components critical for machine learning, including high-quality multiple sequence alignments and insulated training / validation splits that account for deep but only weakly detectable homology across protein space. We have created the ProteinNet series of data sets to provide a standardized mechanism for training and assessing data-driven models of protein sequence-structure relationships. ProteinNet integrates sequence, structure, and evolutionary information in programmatically accessible file formats tailored for machine learning frameworks. Multiple sequence alignments of all structurally characterized proteins were created using substantial high-performance computing resources. Standardized data splits were also generated to emulate the difficulty of past CASP (Critical Assessment of protein Structure Prediction) experiments by resetting protein sequence and structure space to the historical states that preceded six prior CASPs. Utilizing sensitive evolution-based distance metrics to segregate distantly related proteins, we have additionally created validation sets distinct from the official CASP sets that faithfully mimic their difficulty. ProteinNet thus represents a comprehensive and accessible resource for training and assessing machine-learned models of protein structure.