 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHow to Build the Virtual Cell with Artificial Intelligence: Priorities and Opportunities
Sep 18, 2024

The cell is arguably the smallest unit of life and is central to understanding biology. Accurate modeling of cells is important for this understanding as well as for determining the root causes of disease. Recent advances in artificial intelligence (AI), combined with the ability to generate large-scale experimental data, present novel opportunities to model cells. Here we propose a vision of AI-powered Virtual Cells, where robust representations of cells and cellular systems under different conditions are directly learned from growing biological data across measurements and scales. We discuss desired capabilities of AI Virtual Cells, including generating universal representations of biological entities across scales, and facilitating interpretable in silico experiments to predict and understand their behavior using Virtual Instruments. We further address the challenges, opportunities and requirements to realize this vision including data needs, evaluation strategies, and community standards and engagement to ensure biological accuracy and broad utility. We envision a future where AI Virtual Cells help identify new drug targets, predict cellular responses to perturbations, as well as scale hypothesis exploration. With open science collaborations across the biomedical ecosystem that includes academia, philanthropy, and the biopharma and AI industries, a comprehensive predictive understanding of cell mechanisms and interactions is within reach.
BioDiscoveryAgent: An AI Agent for Designing Genetic Perturbation Experiments
May 27, 2024



Agents based on large language models have shown great potential in accelerating scientific discovery by leveraging their rich background knowledge and reasoning capabilities. Here, we develop BioDiscoveryAgent, an agent that designs new experiments, reasons about their outcomes, and efficiently navigates the hypothesis space to reach desired solutions. We demonstrate our agent on the problem of designing genetic perturbation experiments, where the aim is to find a small subset out of many possible genes that, when perturbed, result in a specific phenotype (e.g., cell growth). Utilizing its biological knowledge, BioDiscoveryAgent can uniquely design new experiments without the need to train a machine learning model or explicitly design an acquisition function. Moreover, BioDiscoveryAgent achieves an average of 18% improvement in detecting desired phenotypes across five datasets, compared to existing Bayesian optimization baselines specifically trained for this task. Our evaluation includes one dataset that is unpublished, ensuring it is not part of the language model's training data. Additionally, BioDiscoveryAgent predicts gene combinations to perturb twice as accurately as a random baseline, a task so far not explored in the context of closed-loop experiment design. The agent also has access to tools for searching the biomedical literature, executing code to analyze biological datasets, and prompting another agent to critically evaluate its predictions. Overall, BioDiscoveryAgent is interpretable at every stage, representing an accessible new paradigm in the computational design of biological experiments with the potential to augment scientists' capabilities.
The CausalBench challenge: A machine learning contest for gene network inference from single-cell perturbation data
Aug 29, 2023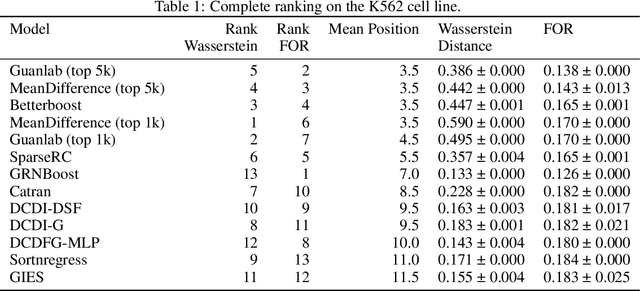
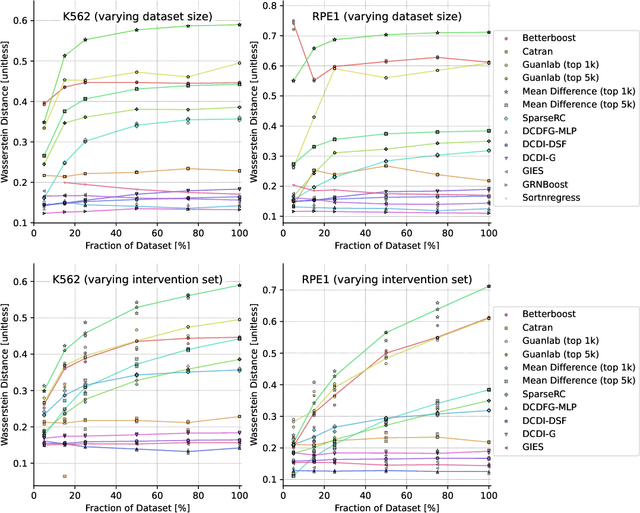
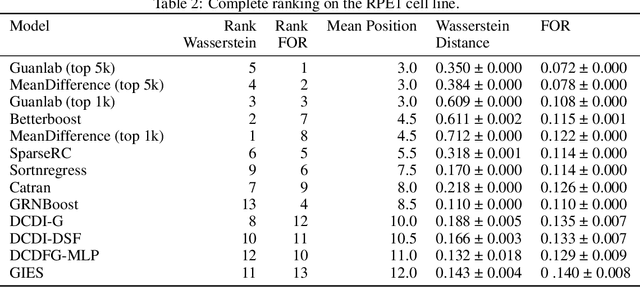
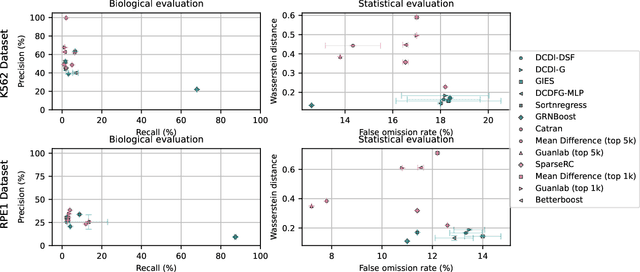
In drug discovery, mapping interactions between genes within cellular systems is a crucial early step. This helps formulate hypotheses regarding molecular mechanisms that could potentially be targeted by future medicines. The CausalBench Challenge was an initiative to invite the machine learning community to advance the state of the art in constructing gene-gene interaction networks. These networks, derived from large-scale, real-world datasets of single cells under various perturbations, are crucial for understanding the causal mechanisms underlying disease biology. Using the framework provided by the CausalBench benchmark, participants were tasked with enhancing the capacity of the state of the art methods to leverage large-scale genetic perturbation data. This report provides an analysis and summary of the methods submitted during the challenge to give a partial image of the state of the art at the time of the challenge. The winning solutions significantly improved performance compared to previous baselines, establishing a new state of the art for this critical task in biology and medicine.
Zero-shot causal learning
Jan 28, 2023Predicting how different interventions will causally affect a specific individual is important in a variety of domains such as personalized medicine, public policy, and online marketing. However, most existing causal methods cannot generalize to predicting the effects of previously unseen interventions (e.g., a newly invented drug), because they require data for individuals who received the intervention. Here, we consider zero-shot causal learning: predicting the personalized effects of novel, previously unseen interventions. To tackle this problem, we propose CaML, a causal meta-learning framework which formulates the personalized prediction of each intervention's effect as a task. Rather than training a separate model for each intervention, CaML trains as a single meta-model across thousands of tasks, each constructed by sampling an intervention and individuals who either did or did not receive it. By leveraging both intervention information (e.g., a drug's attributes) and individual features (e.g., a patient's history), CaML is able to predict the personalized effects of unseen interventions. Experimental results on real world datasets in large-scale medical claims and cell-line perturbations demonstrate the effectiveness of our approach. Most strikingly, CaML zero-shot predictions outperform even strong baselines which have direct access to data of considered target interventions.
CausalBench: A Large-scale Benchmark for Network Inference from Single-cell Perturbation Data
Oct 31, 2022

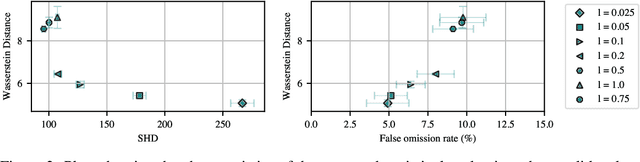

Mapping biological mechanisms in cellular systems is a fundamental step in early-stage drug discovery that serves to generate hypotheses on what disease-relevant molecular targets may effectively be modulated by pharmacological interventions. With the advent of high-throughput methods for measuring single-cell gene expression under genetic perturbations, we now have effective means for generating evidence for causal gene-gene interactions at scale. However, inferring graphical networks of the size typically encountered in real-world gene-gene interaction networks is difficult in terms of both achieving and evaluating faithfulness to the true underlying causal graph. Moreover, standardised benchmarks for comparing methods for causal discovery in perturbational single-cell data do not yet exist. Here, we introduce CausalBench - a comprehensive benchmark suite for evaluating network inference methods on large-scale perturbational single-cell gene expression data. CausalBench introduces several biologically meaningful performance metrics and operates on two large, curated and openly available benchmark data sets for evaluating methods on the inference of gene regulatory networks from single-cell data generated under perturbations. With real-world datasets consisting of over \numprint{200000} training samples under interventions, CausalBench could potentially help facilitate advances in causal network inference by providing what is - to the best of our knowledge - the largest openly available test bed for causal discovery from real-world perturbation data to date.
On the Opportunities and Risks of Foundation Models
Aug 18, 2021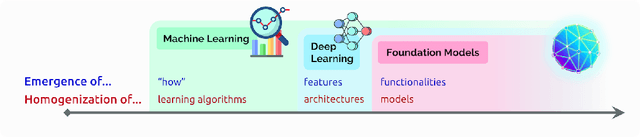
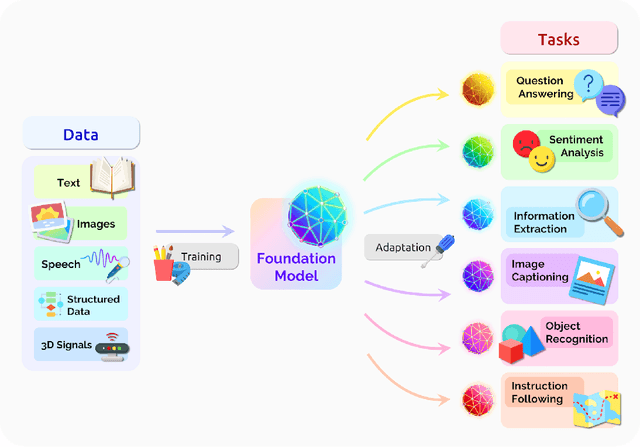
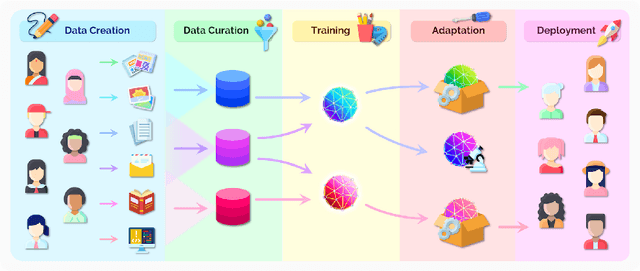
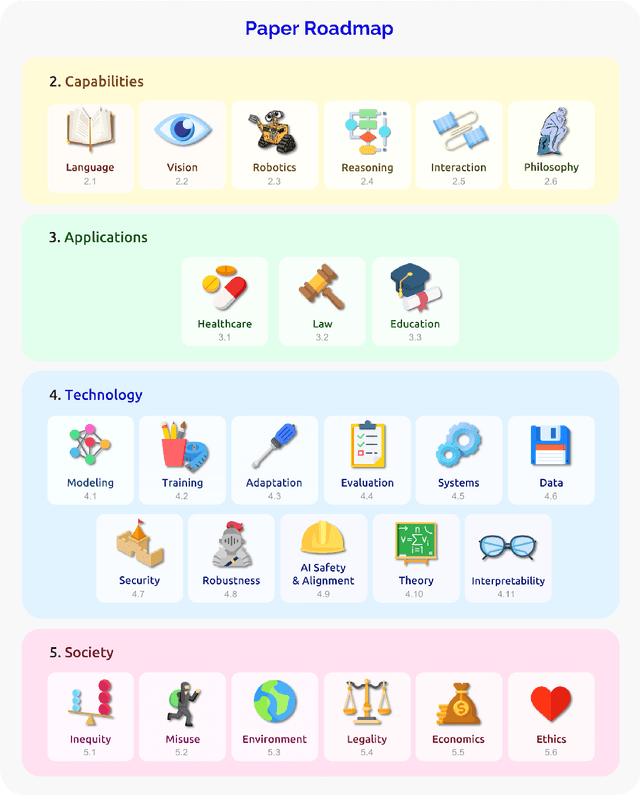
AI is undergoing a paradigm shift with the rise of models (e.g., BERT, DALL-E, GPT-3) that are trained on broad data at scale and are adaptable to a wide range of downstream tasks. We call these models foundation models to underscore their critically central yet incomplete character. This report provides a thorough account of the opportunities and risks of foundation models, ranging from their capabilities (e.g., language, vision, robotics, reasoning, human interaction) and technical principles(e.g., model architectures, training procedures, data, systems, security, evaluation, theory) to their applications (e.g., law, healthcare, education) and societal impact (e.g., inequity, misuse, economic and environmental impact, legal and ethical considerations). Though foundation models are based on standard deep learning and transfer learning, their scale results in new emergent capabilities,and their effectiveness across so many tasks incentivizes homogenization. Homogenization provides powerful leverage but demands caution, as the defects of the foundation model are inherited by all the adapted models downstream. Despite the impending widespread deployment of foundation models, we currently lack a clear understanding of how they work, when they fail, and what they are even capable of due to their emergent properties. To tackle these questions, we believe much of the critical research on foundation models will require deep interdisciplinary collaboration commensurate with their fundamentally sociotechnical nature.
Therapeutics Data Commons: Machine Learning Datasets and Tasks for Therapeutics
Feb 18, 2021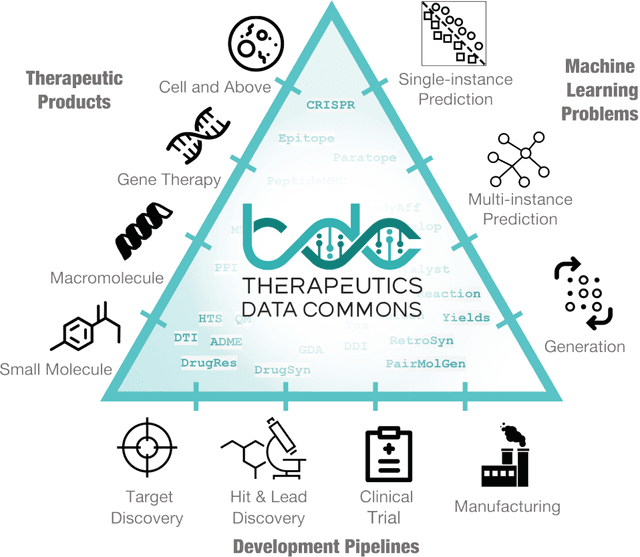
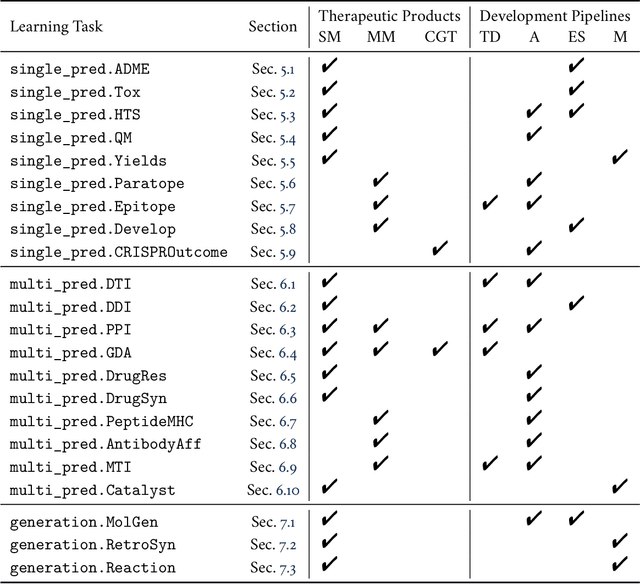
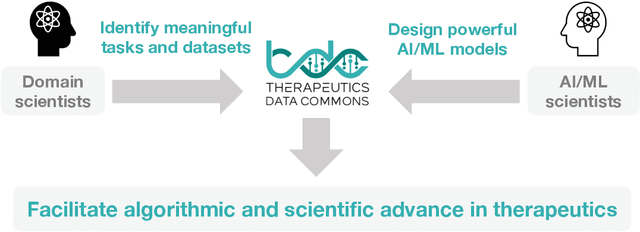
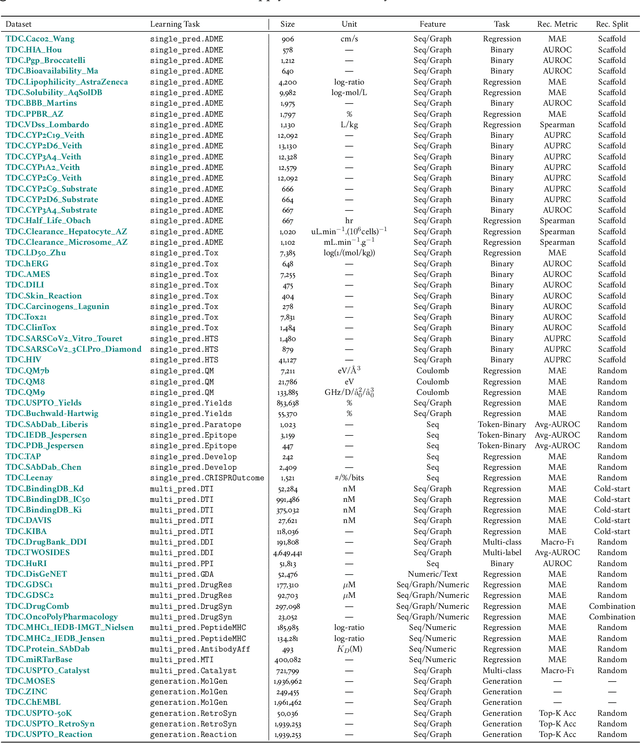
Machine learning for therapeutics is an emerging field with incredible opportunities for innovation and expansion. Despite the initial success, many key challenges remain open. Here, we introduce Therapeutics Data Commons (TDC), the first unifying framework to systematically access and evaluate machine learning across the entire range of therapeutics. At its core, TDC is a collection of curated datasets and learning tasks that can translate algorithmic innovation into biomedical and clinical implementation. To date, TDC includes 66 machine learning-ready datasets from 22 learning tasks, spanning the discovery and development of safe and effective medicines. TDC also provides an ecosystem of tools, libraries, leaderboards, and community resources, including data functions, strategies for systematic model evaluation, meaningful data splits, data processors, and molecule generation oracles. All datasets and learning tasks are integrated and accessible via an open-source library. We envision that TDC can facilitate algorithmic and scientific advances and accelerate development, validation, and transition into production and clinical implementation. TDC is a continuous, open-source initiative, and we invite contributions from the research community. TDC is publicly available at https://tdcommons.ai.