 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIn-silico biological discovery with large perturbation models
Mar 30, 2025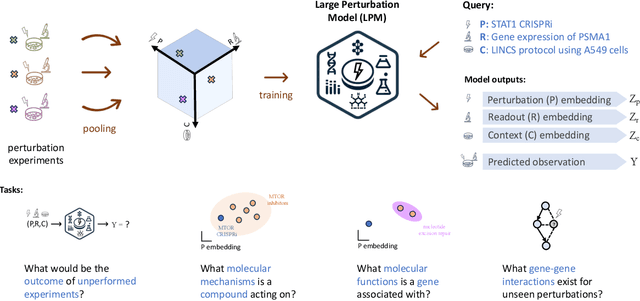
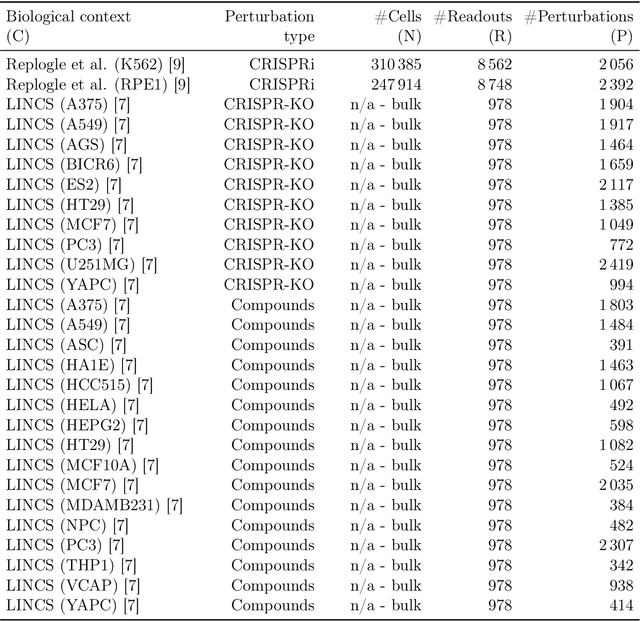
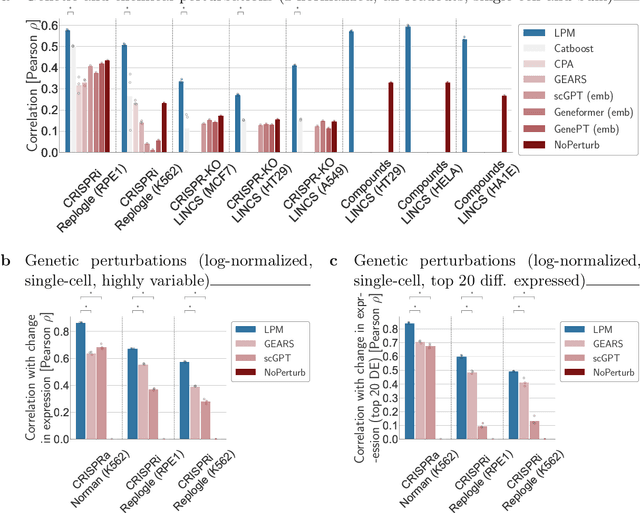
Data generated in perturbation experiments link perturbations to the changes they elicit and therefore contain information relevant to numerous biological discovery tasks -- from understanding the relationships between biological entities to developing therapeutics. However, these data encompass diverse perturbations and readouts, and the complex dependence of experimental outcomes on their biological context makes it challenging to integrate insights across experiments. Here, we present the Large Perturbation Model (LPM), a deep-learning model that integrates multiple, heterogeneous perturbation experiments by representing perturbation, readout, and context as disentangled dimensions. LPM outperforms existing methods across multiple biological discovery tasks, including in predicting post-perturbation transcriptomes of unseen experiments, identifying shared molecular mechanisms of action between chemical and genetic perturbations, and facilitating the inference of gene-gene interaction networks.
Multi-megabase scale genome interpretation with genetic language models
Jan 13, 2025Understanding how molecular changes caused by genetic variation drive disease risk is crucial for deciphering disease mechanisms. However, interpreting genome sequences is challenging because of the vast size of the human genome, and because its consequences manifest across a wide range of cells, tissues and scales -- spanning from molecular to whole organism level. Here, we present Phenformer, a multi-scale genetic language model that learns to generate mechanistic hypotheses as to how differences in genome sequence lead to disease-relevant changes in expression across cell types and tissues directly from DNA sequences of up to 88 million base pairs. Using whole genome sequencing data from more than 150 000 individuals, we show that Phenformer generates mechanistic hypotheses about disease-relevant cell and tissue types that match literature better than existing state-of-the-art methods, while using only sequence data. Furthermore, disease risk predictors enriched by Phenformer show improved prediction performance and generalisation to diverse populations. Accurate multi-megabase scale interpretation of whole genomes without additional experimental data enables both a deeper understanding of molecular mechanisms involved in disease and improved disease risk prediction at the level of individuals.
Efficient Differentiable Discovery of Causal Order
Oct 11, 2024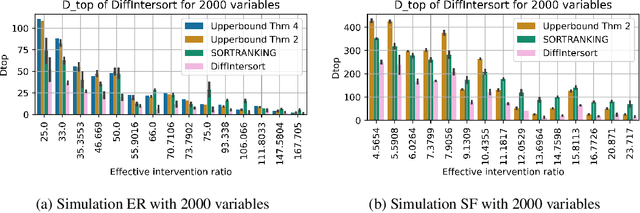
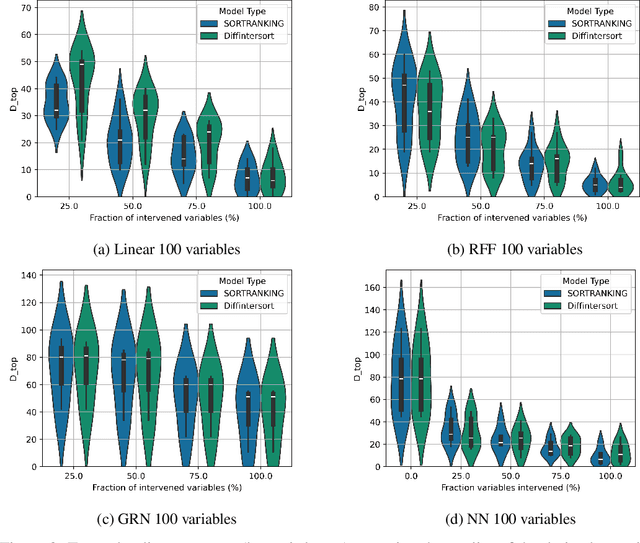
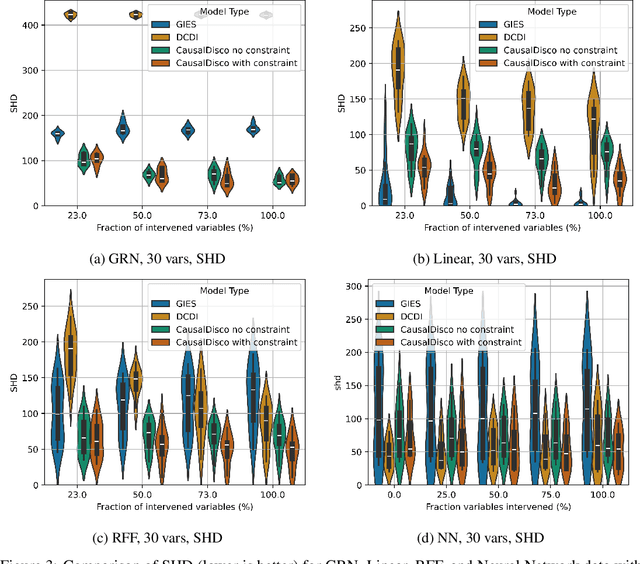
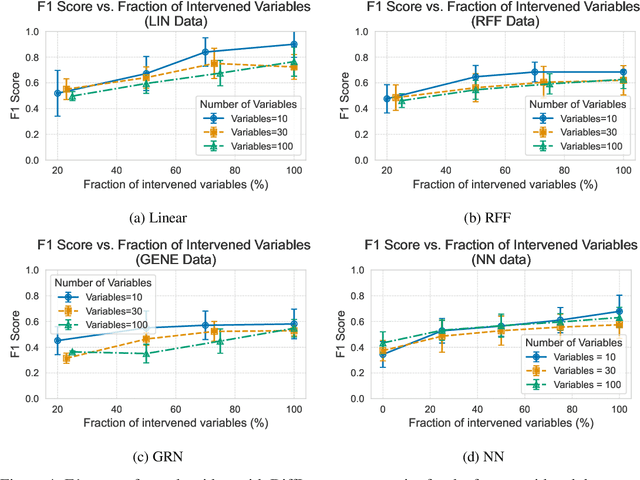
In the algorithm Intersort, Chevalley et al. (2024) proposed a score-based method to discover the causal order of variables in a Directed Acyclic Graph (DAG) model, leveraging interventional data to outperform existing methods. However, as a score-based method over the permutahedron, Intersort is computationally expensive and non-differentiable, limiting its ability to be utilised in problems involving large-scale datasets, such as those in genomics and climate models, or to be integrated into end-to-end gradient-based learning frameworks. We address this limitation by reformulating Intersort using differentiable sorting and ranking techniques. Our approach enables scalable and differentiable optimization of causal orderings, allowing the continuous score function to be incorporated as a regularizer in downstream tasks. Empirical results demonstrate that causal discovery algorithms benefit significantly from regularizing on the causal order, underscoring the effectiveness of our method. Our work opens the door to efficiently incorporating regularization for causal order into the training of differentiable models and thereby addresses a long-standing limitation of purely associational supervised learning.
Deriving Causal Order from Single-Variable Interventions: Guarantees & Algorithm
May 28, 2024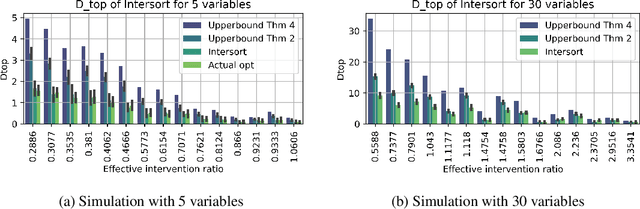
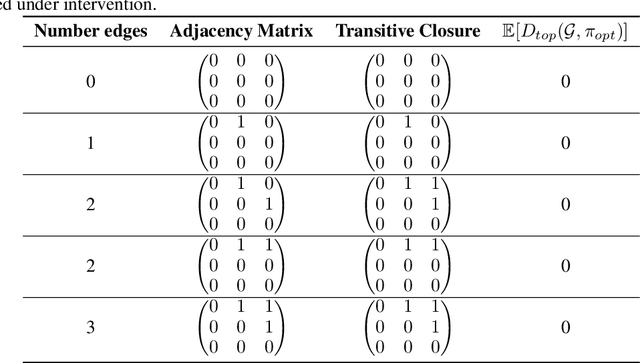
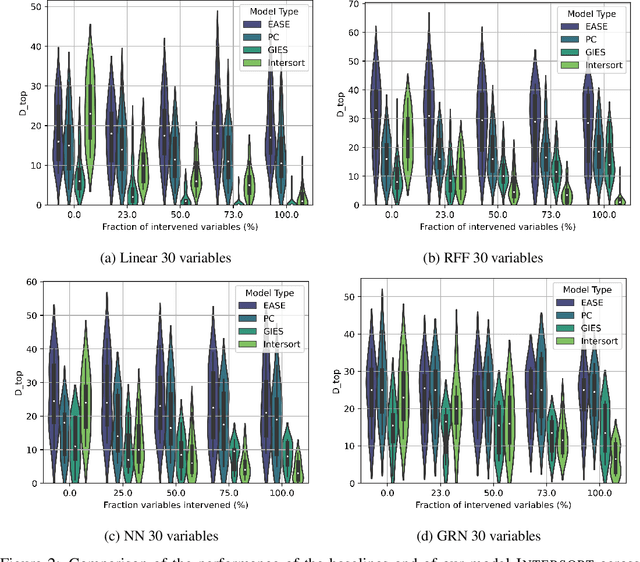
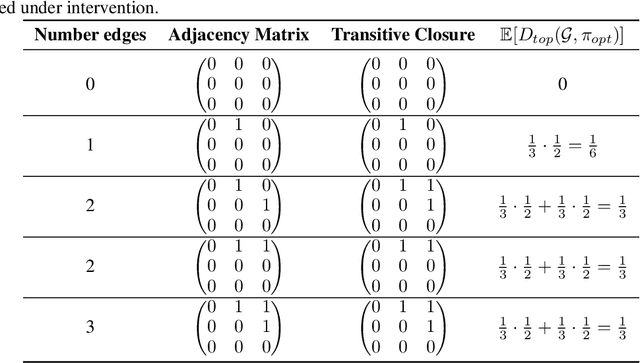
Targeted and uniform interventions to a system are crucial for unveiling causal relationships. While several methods have been developed to leverage interventional data for causal structure learning, their practical application in real-world scenarios often remains challenging. Recent benchmark studies have highlighted these difficulties, even when large numbers of single-variable intervention samples are available. In this work, we demonstrate, both theoretically and empirically, that such datasets contain a wealth of causal information that can be effectively extracted under realistic assumptions about the data distribution. More specifically, we introduce the notion of interventional faithfulness, which relies on comparisons between the marginal distributions of each variable across observational and interventional settings, and we introduce a score on causal orders. Under this assumption, we are able to prove strong theoretical guarantees on the optimum of our score that also hold for large-scale settings. To empirically verify our theory, we introduce Intersort, an algorithm designed to infer the causal order from datasets containing large numbers of single-variable interventions by approximately optimizing our score. Intersort outperforms baselines (GIES, PC and EASE) on almost all simulated data settings replicating common benchmarks in the field. Our proposed novel approach to modeling interventional datasets thus offers a promising avenue for advancing causal inference, highlighting significant potential for further enhancements under realistic assumptions.
Sample Selection Bias in Machine Learning for Healthcare
May 13, 2024



While machine learning algorithms hold promise for personalised medicine, their clinical adoption remains limited. One critical factor contributing to this restraint is sample selection bias (SSB) which refers to the study population being less representative of the target population, leading to biased and potentially harmful decisions. Despite being well-known in the literature, SSB remains scarcely studied in machine learning for healthcare. Moreover, the existing techniques try to correct the bias by balancing distributions between the study and the target populations, which may result in a loss of predictive performance. To address these problems, our study illustrates the potential risks associated with SSB by examining SSB's impact on the performance of machine learning algorithms. Most importantly, we propose a new research direction for addressing SSB, based on the target population identification rather than the bias correction. Specifically, we propose two independent networks (T-Net) and a multitasking network (MT-Net) for addressing SSB, where one network/task identifies the target subpopulation which is representative of the study population and the second makes predictions for the identified subpopulation. Our empirical results with synthetic and semi-synthetic datasets highlight that SSB can lead to a large drop in the performance of an algorithm for the target population as compared with the study population, as well as a substantial difference in the performance for the target subpopulations that are representative of the selected and the non-selected patients from the study population. Furthermore, our proposed techniques demonstrate robustness across various settings, including different dataset sizes, event rates, and selection rates, outperforming the existing bias correction techniques.
A collection of the accepted papers for the Human-Centric Representation Learning workshop at AAAI 2024
Mar 14, 2024This non-archival index is not complete, as some accepted papers chose to opt-out of inclusion. The list of all accepted papers is available on the workshop website.
DiscoBAX: Discovery of Optimal Intervention Sets in Genomic Experiment Design
Dec 07, 2023The discovery of therapeutics to treat genetically-driven pathologies relies on identifying genes involved in the underlying disease mechanisms. Existing approaches search over the billions of potential interventions to maximize the expected influence on the target phenotype. However, to reduce the risk of failure in future stages of trials, practical experiment design aims to find a set of interventions that maximally change a target phenotype via diverse mechanisms. We propose DiscoBAX, a sample-efficient method for maximizing the rate of significant discoveries per experiment while simultaneously probing for a wide range of diverse mechanisms during a genomic experiment campaign. We provide theoretical guarantees of approximate optimality under standard assumptions, and conduct a comprehensive experimental evaluation covering both synthetic as well as real-world experimental design tasks. DiscoBAX outperforms existing state-of-the-art methods for experimental design, selecting effective and diverse perturbations in biological systems.
The CausalBench challenge: A machine learning contest for gene network inference from single-cell perturbation data
Aug 29, 2023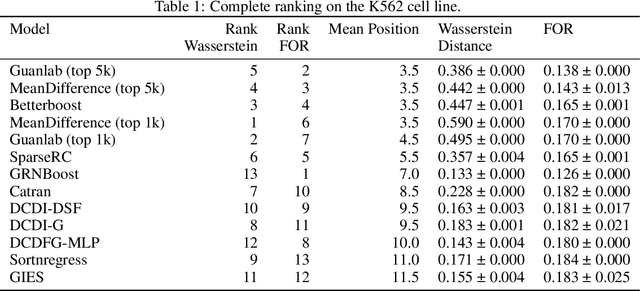
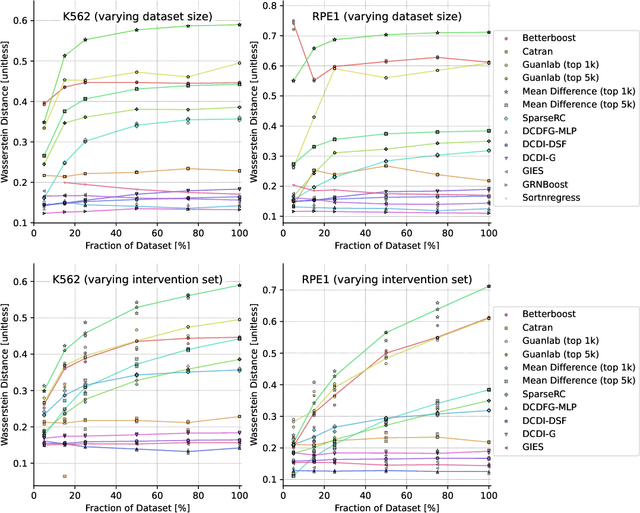
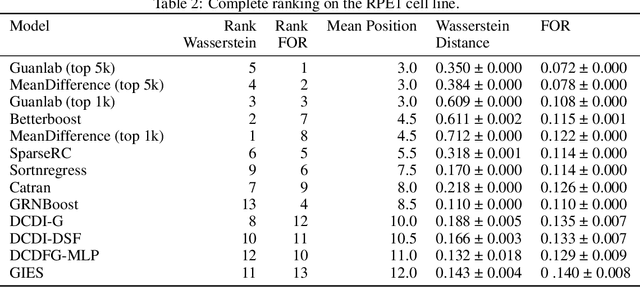
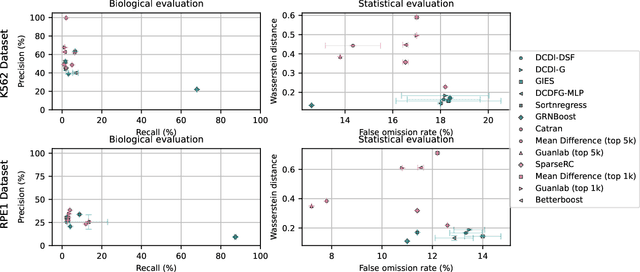
In drug discovery, mapping interactions between genes within cellular systems is a crucial early step. This helps formulate hypotheses regarding molecular mechanisms that could potentially be targeted by future medicines. The CausalBench Challenge was an initiative to invite the machine learning community to advance the state of the art in constructing gene-gene interaction networks. These networks, derived from large-scale, real-world datasets of single cells under various perturbations, are crucial for understanding the causal mechanisms underlying disease biology. Using the framework provided by the CausalBench benchmark, participants were tasked with enhancing the capacity of the state of the art methods to leverage large-scale genetic perturbation data. This report provides an analysis and summary of the methods submitted during the challenge to give a partial image of the state of the art at the time of the challenge. The winning solutions significantly improved performance compared to previous baselines, establishing a new state of the art for this critical task in biology and medicine.
Multi-omics Prediction from High-content Cellular Imaging with Deep Learning
Jun 19, 2023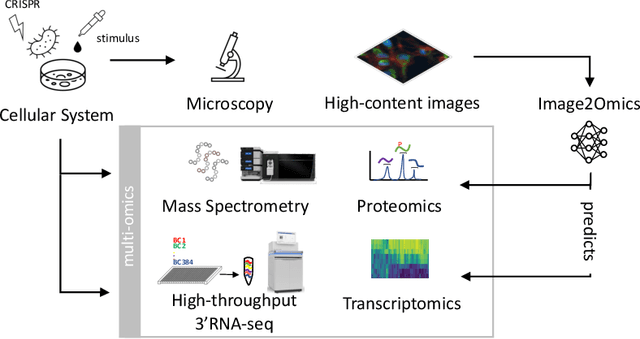
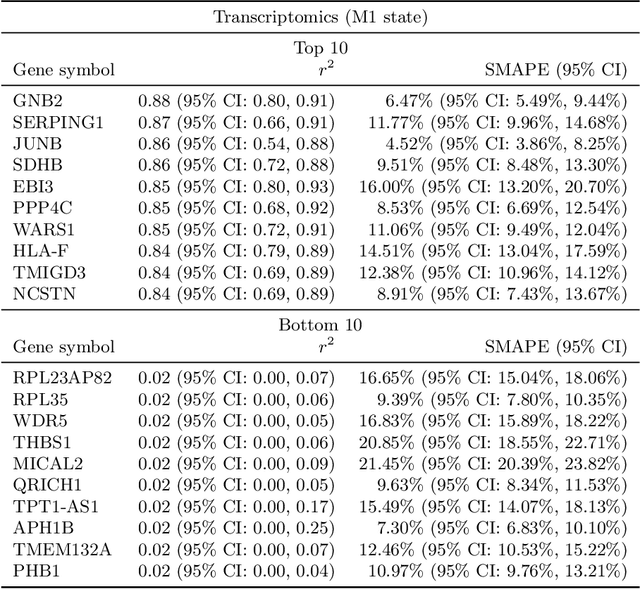
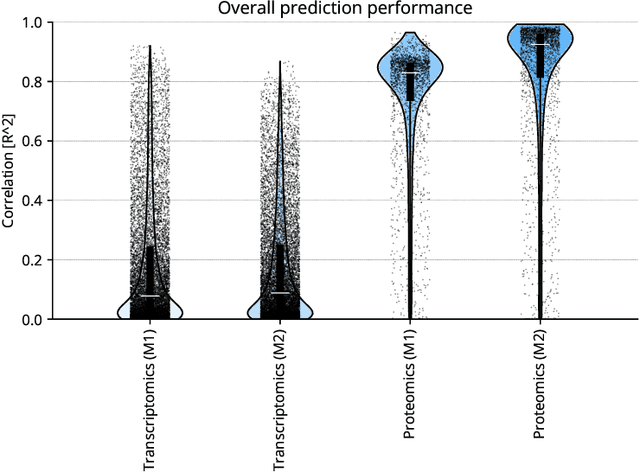
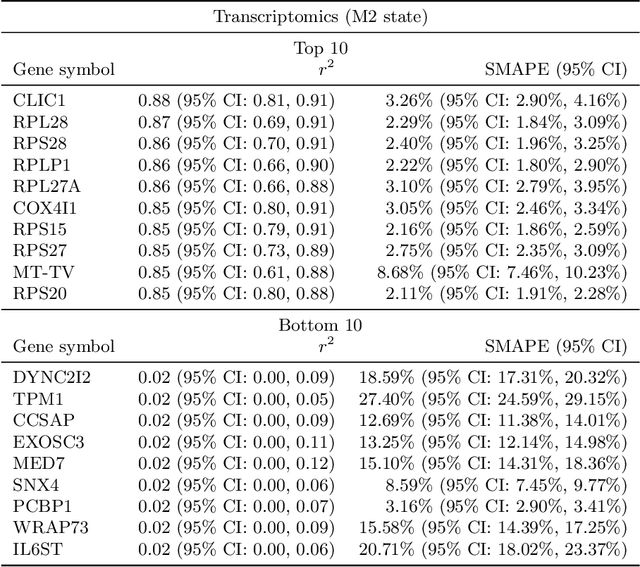
High-content cellular imaging, transcriptomics, and proteomics data provide rich and complementary views on the molecular layers of biology that influence cellular states and function. However, the biological determinants through which changes in multi-omics measurements influence cellular morphology have not yet been systematically explored, and the degree to which cell imaging could potentially enable the prediction of multi-omics directly from cell imaging data is therefore currently unclear. Here, we address the question of whether it is possible to predict bulk multi-omics measurements directly from cell images using Image2Omics -- a deep learning approach that predicts multi-omics in a cell population directly from high-content images stained with multiplexed fluorescent dyes. We perform an experimental evaluation in gene-edited macrophages derived from human induced pluripotent stem cell (hiPSC) under multiple stimulation conditions and demonstrate that Image2Omics achieves significantly better performance in predicting transcriptomics and proteomics measurements directly from cell images than predictors based on the mean observed training set abundance. We observed significant predictability of abundances for 5903 (22.43%; 95% CI: 8.77%, 38.88%) and 5819 (22.11%; 95% CI: 10.40%, 38.08%) transcripts out of 26137 in M1 and M2-stimulated macrophages respectively and for 1933 (38.77%; 95% CI: 36.94%, 39.85%) and 2055 (41.22%; 95% CI: 39.31%, 42.42%) proteins out of 4986 in M1 and M2-stimulated macrophages respectively. Our results show that some transcript and protein abundances are predictable from cell imaging and that cell imaging may potentially, in some settings and depending on the mechanisms of interest and desired performance threshold, even be a scalable and resource-efficient substitute for multi-omics measurements.
FED-CD: Federated Causal Discovery from Interventional and Observational Data
Nov 07, 2022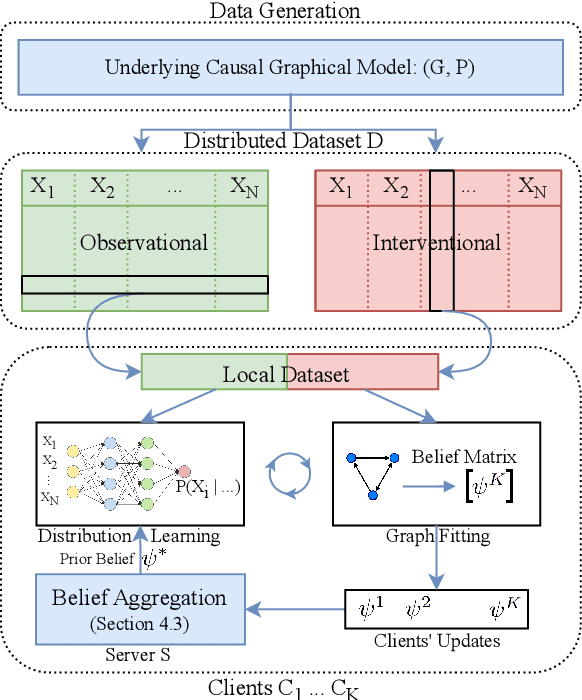
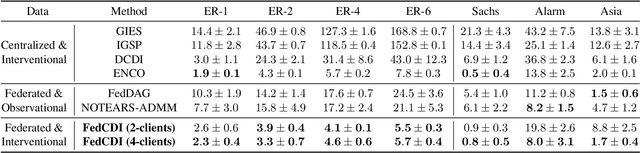
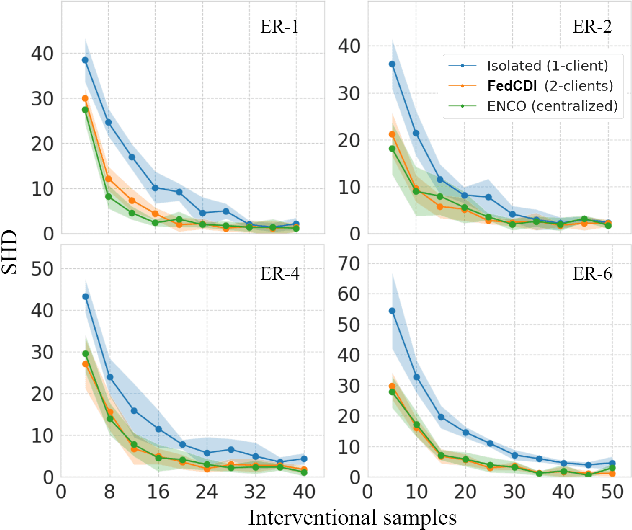

Causal discovery, the inference of causal relations from data, is a core task of fundamental importance in all scientific domains, and several new machine learning methods for addressing the causal discovery problem have been proposed recently. However, existing machine learning methods for causal discovery typically require that the data used for inference is pooled and available in a centralized location. In many domains of high practical importance, such as in healthcare, data is only available at local data-generating entities (e.g. hospitals in the healthcare context), and cannot be shared across entities due to, among others, privacy and regulatory reasons. In this work, we address the problem of inferring causal structure - in the form of a directed acyclic graph (DAG) - from a distributed data set that contains both observational and interventional data in a privacy-preserving manner by exchanging updates instead of samples. To this end, we introduce a new federated framework, FED-CD, that enables the discovery of global causal structures both when the set of intervened covariates is the same across decentralized entities, and when the set of intervened covariates are potentially disjoint. We perform a comprehensive experimental evaluation on synthetic data that demonstrates that FED-CD enables effective aggregation of decentralized data for causal discovery without direct sample sharing, even when the contributing distributed data sets cover disjoint sets of interventions. Effective methods for causal discovery in distributed data sets could significantly advance scientific discovery and knowledge sharing in important settings, for instance, healthcare, in which sharing of data across local sites is difficult or prohibited.