 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMulti-omics Prediction from High-content Cellular Imaging with Deep Learning
Jun 19, 2023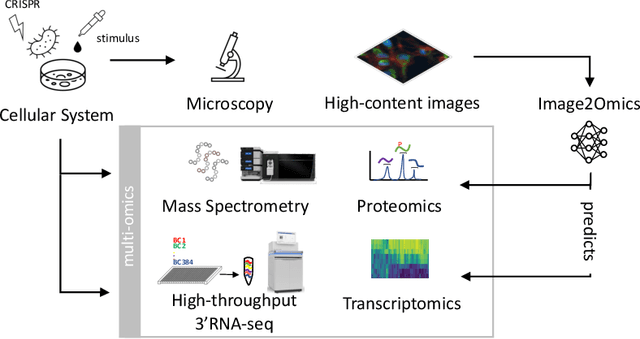
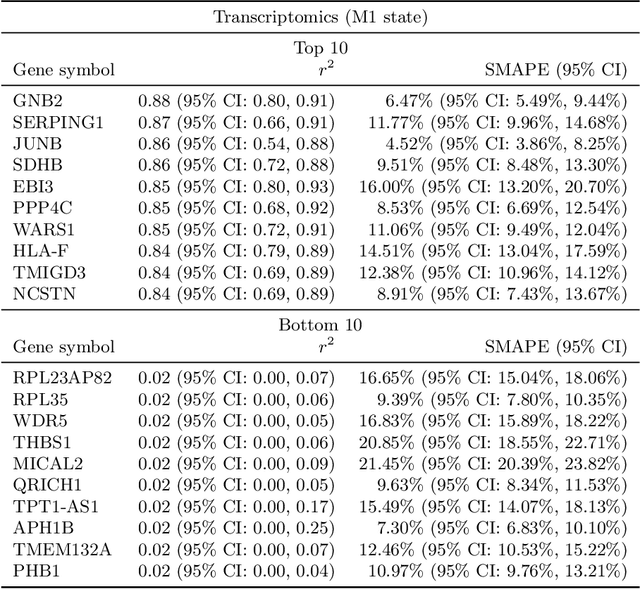
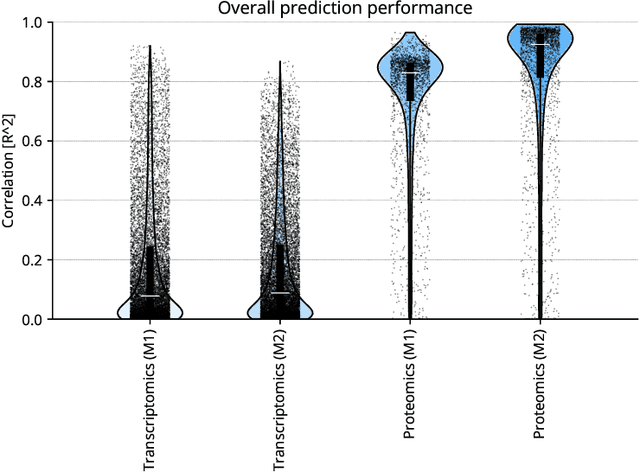
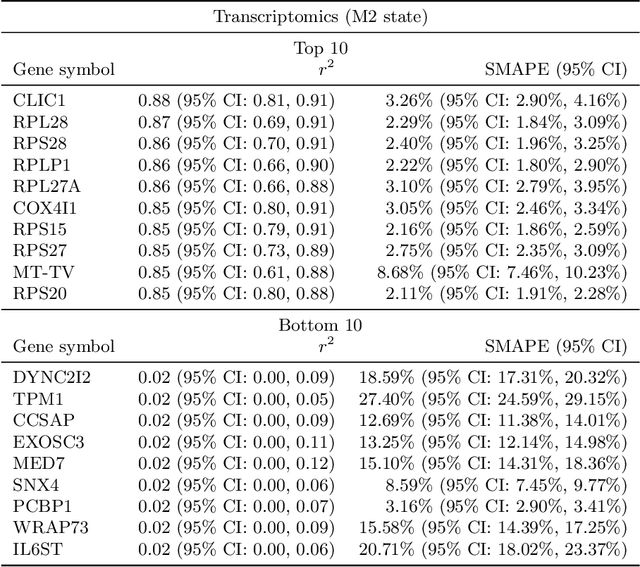
High-content cellular imaging, transcriptomics, and proteomics data provide rich and complementary views on the molecular layers of biology that influence cellular states and function. However, the biological determinants through which changes in multi-omics measurements influence cellular morphology have not yet been systematically explored, and the degree to which cell imaging could potentially enable the prediction of multi-omics directly from cell imaging data is therefore currently unclear. Here, we address the question of whether it is possible to predict bulk multi-omics measurements directly from cell images using Image2Omics -- a deep learning approach that predicts multi-omics in a cell population directly from high-content images stained with multiplexed fluorescent dyes. We perform an experimental evaluation in gene-edited macrophages derived from human induced pluripotent stem cell (hiPSC) under multiple stimulation conditions and demonstrate that Image2Omics achieves significantly better performance in predicting transcriptomics and proteomics measurements directly from cell images than predictors based on the mean observed training set abundance. We observed significant predictability of abundances for 5903 (22.43%; 95% CI: 8.77%, 38.88%) and 5819 (22.11%; 95% CI: 10.40%, 38.08%) transcripts out of 26137 in M1 and M2-stimulated macrophages respectively and for 1933 (38.77%; 95% CI: 36.94%, 39.85%) and 2055 (41.22%; 95% CI: 39.31%, 42.42%) proteins out of 4986 in M1 and M2-stimulated macrophages respectively. Our results show that some transcript and protein abundances are predictable from cell imaging and that cell imaging may potentially, in some settings and depending on the mechanisms of interest and desired performance threshold, even be a scalable and resource-efficient substitute for multi-omics measurements.
Automating Whole Brain Histology to MRI Registration: Implementation of a Computational Pipeline
May 22, 2019

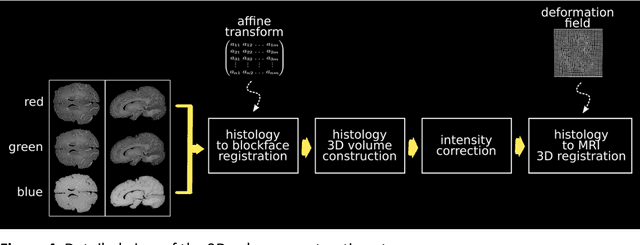
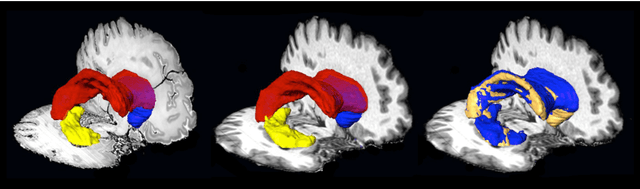
Although the latest advances in MRI technology have allowed the acquisition of higher resolution images, reliable delineation of cytoarchitectural or subcortical nuclei boundaries is not possible. As a result, histological images are still required to identify the exact limits of neuroanatomical structures. However, histological processing is associated with tissue distortion and fixation artifacts, which prevent a direct comparison between the two modalities. Our group has previously proposed a histological procedure based on celloidin embedding that reduces the amount of artifacts and yields high quality whole brain histological slices. Celloidin embedded tissue, nevertheless, still bears distortions that must be corrected. We propose a computational pipeline designed to semi-automatically process the celloidin embedded histology and register them to their MRI counterparts. In this paper we report the accuracy of our pipeline in two whole brain volumes from the Brain Bank of the Brazilian Aging Brain Study Group (BBBABSG). Results were assessed by comparison of manual segmentations from two experts in both MRIs and the registered histological volumes. The two whole brain histology/MRI datasets were successfully registered using minimal user interaction. We also point to possible improvements based on recent implementations that could be added to this pipeline, potentially allowing for higher precision and further performance gains.