 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMulti-megabase scale genome interpretation with genetic language models
Jan 13, 2025Understanding how molecular changes caused by genetic variation drive disease risk is crucial for deciphering disease mechanisms. However, interpreting genome sequences is challenging because of the vast size of the human genome, and because its consequences manifest across a wide range of cells, tissues and scales -- spanning from molecular to whole organism level. Here, we present Phenformer, a multi-scale genetic language model that learns to generate mechanistic hypotheses as to how differences in genome sequence lead to disease-relevant changes in expression across cell types and tissues directly from DNA sequences of up to 88 million base pairs. Using whole genome sequencing data from more than 150 000 individuals, we show that Phenformer generates mechanistic hypotheses about disease-relevant cell and tissue types that match literature better than existing state-of-the-art methods, while using only sequence data. Furthermore, disease risk predictors enriched by Phenformer show improved prediction performance and generalisation to diverse populations. Accurate multi-megabase scale interpretation of whole genomes without additional experimental data enables both a deeper understanding of molecular mechanisms involved in disease and improved disease risk prediction at the level of individuals.
Sample Selection Bias in Machine Learning for Healthcare
May 13, 2024



While machine learning algorithms hold promise for personalised medicine, their clinical adoption remains limited. One critical factor contributing to this restraint is sample selection bias (SSB) which refers to the study population being less representative of the target population, leading to biased and potentially harmful decisions. Despite being well-known in the literature, SSB remains scarcely studied in machine learning for healthcare. Moreover, the existing techniques try to correct the bias by balancing distributions between the study and the target populations, which may result in a loss of predictive performance. To address these problems, our study illustrates the potential risks associated with SSB by examining SSB's impact on the performance of machine learning algorithms. Most importantly, we propose a new research direction for addressing SSB, based on the target population identification rather than the bias correction. Specifically, we propose two independent networks (T-Net) and a multitasking network (MT-Net) for addressing SSB, where one network/task identifies the target subpopulation which is representative of the study population and the second makes predictions for the identified subpopulation. Our empirical results with synthetic and semi-synthetic datasets highlight that SSB can lead to a large drop in the performance of an algorithm for the target population as compared with the study population, as well as a substantial difference in the performance for the target subpopulations that are representative of the selected and the non-selected patients from the study population. Furthermore, our proposed techniques demonstrate robustness across various settings, including different dataset sizes, event rates, and selection rates, outperforming the existing bias correction techniques.
Generalising sequence models for epigenome predictions with tissue and assay embeddings
Aug 22, 2023Sequence modelling approaches for epigenetic profile prediction have recently expanded in terms of sequence length, model size, and profile diversity. However, current models cannot infer on many experimentally feasible tissue and assay pairs due to poor usage of contextual information, limiting $\textit{in silico}$ understanding of regulatory genomics. We demonstrate that strong correlation can be achieved across a large range of experimental conditions by integrating tissue and assay embeddings into a Contextualised Genomic Network (CGN). In contrast to previous approaches, we enhance long-range sequence embeddings with contextual information in the input space, rather than expanding the output space. We exhibit the efficacy of our approach across a broad set of epigenetic profiles and provide the first insights into the effect of genetic variants on epigenetic sequence model training. Our general approach to context integration exceeds state of the art in multiple settings while employing a more rigorous validation procedure.
ALBRT: Cellular Composition Prediction in Routine Histology Images
Aug 26, 2021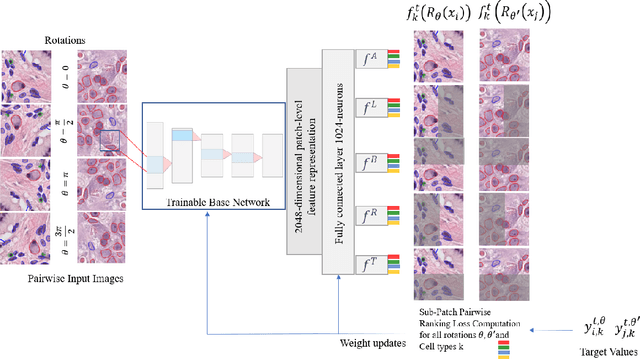
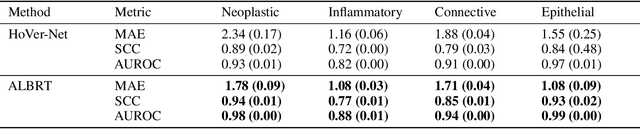
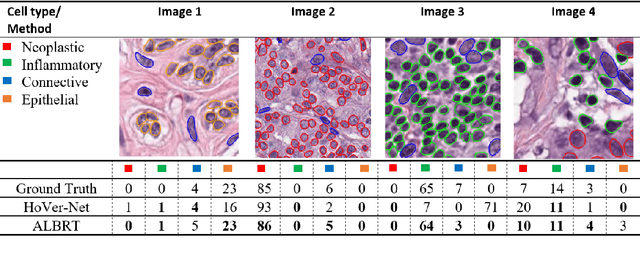
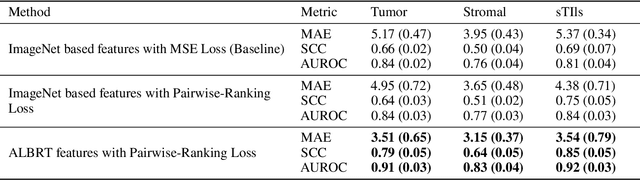
Cellular composition prediction, i.e., predicting the presence and counts of different types of cells in the tumor microenvironment from a digitized image of a Hematoxylin and Eosin (H&E) stained tissue section can be used for various tasks in computational pathology such as the analysis of cellular topology and interactions, subtype prediction, survival analysis, etc. In this work, we propose an image-based cellular composition predictor (ALBRT) which can accurately predict the presence and counts of different types of cells in a given image patch. ALBRT, by its contrastive-learning inspired design, learns a compact and rotation-invariant feature representation that is then used for cellular composition prediction of different cell types. It offers significant improvement over existing state-of-the-art approaches for cell classification and counting. The patch-level feature representation learned by ALBRT is transferrable for cellular composition analysis over novel datasets and can also be utilized for downstream prediction tasks in CPath as well. The code and the inference webserver for the proposed method are available at the URL: https://github.com/engrodawood/ALBRT.
All You Need is Color: Image based Spatial Gene Expression Prediction using Neural Stain Learning
Aug 26, 2021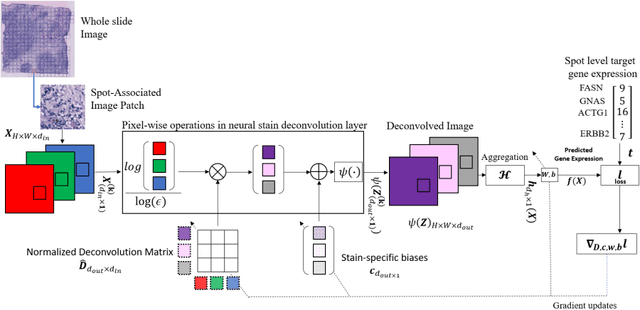
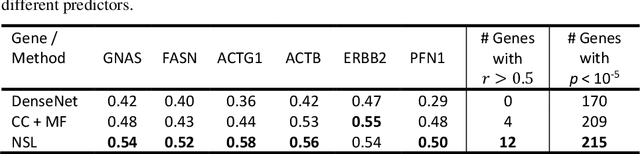
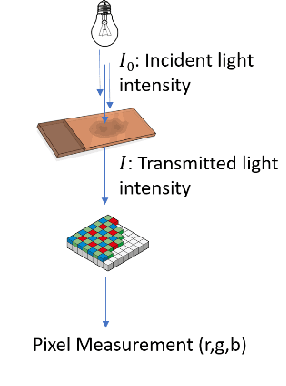
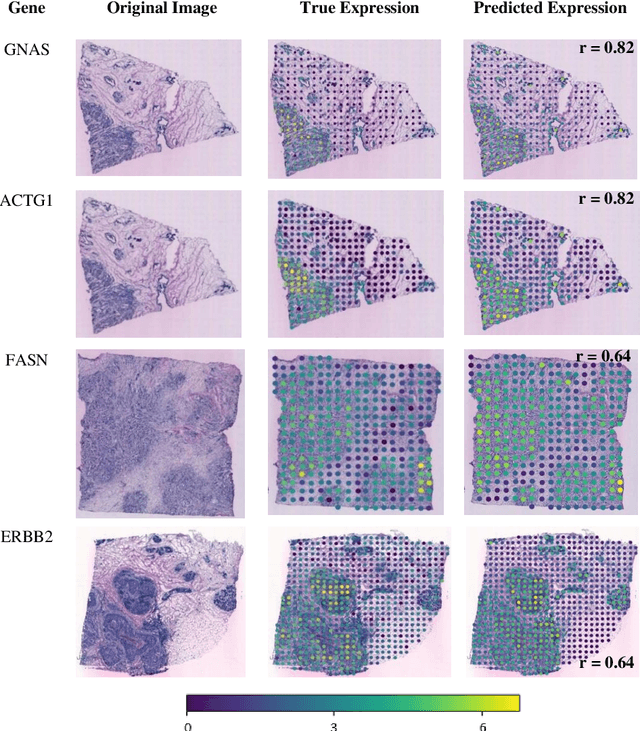
"Is it possible to predict expression levels of different genes at a given spatial location in the routine histology image of a tumor section by modeling its stain absorption characteristics?" In this work, we propose a "stain-aware" machine learning approach for prediction of spatial transcriptomic gene expression profiles using digital pathology image of a routine Hematoxylin & Eosin (H&E) histology section. Unlike recent deep learning methods which are used for gene expression prediction, our proposed approach termed Neural Stain Learning (NSL) explicitly models the association of stain absorption characteristics of the tissue with gene expression patterns in spatial transcriptomics by learning a problem-specific stain deconvolution matrix in an end-to-end manner. The proposed method with only 11 trainable weight parameters outperforms both classical regression models with cellular composition and morphological features as well as deep learning methods. We have found that the gene expression predictions from the proposed approach show higher correlations with true expression values obtained through sequencing for a larger set of genes in comparison to other approaches.
Consistency of mechanistic causal discovery in continuous-time using Neural ODEs
May 06, 2021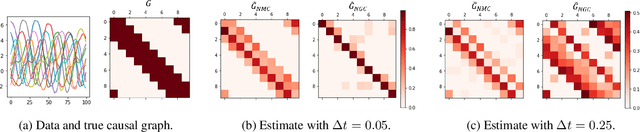
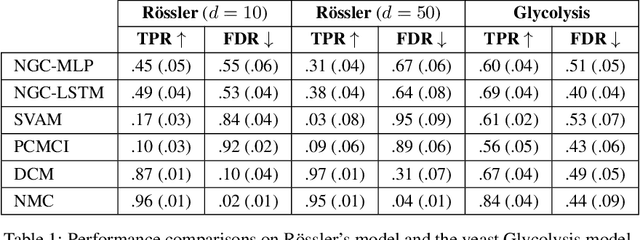
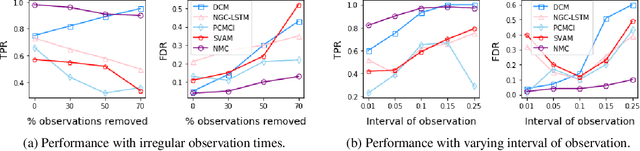

The discovery of causal mechanisms from time series data is a key problem in fields working with complex systems. Most identifiability results and learning algorithms assume the underlying dynamics to be discrete in time. Comparatively few, in contrast, explicitly define causal associations in infinitesimal intervals of time, independently of the scale of observation and of the regularity of sampling. In this paper, we consider causal discovery in continuous-time for the study of dynamical systems. We prove that for vector fields parameterized in a large class of neural networks, adaptive regularization schemes consistently recover causal graphs in systems of ordinary differential equations (ODEs). Using this insight, we propose a causal discovery algorithm based on penalized Neural ODEs that we show to be applicable to the general setting of irregularly-sampled multivariate time series and to strongly outperform the state of the art.