 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Novel Approach to Linking Histology Images with DNA Methylation
Apr 07, 2025DNA methylation is an epigenetic mechanism that regulates gene expression by adding methyl groups to DNA. Abnormal methylation patterns can disrupt gene expression and have been linked to cancer development. To quantify DNA methylation, specialized assays are typically used. However, these assays are often costly and have lengthy processing times, which limits their widespread availability in routine clinical practice. In contrast, whole slide images (WSIs) for the majority of cancer patients can be more readily available. As such, given the ready availability of WSIs, there is a compelling need to explore the potential relationship between WSIs and DNA methylation patterns. To address this, we propose an end-to-end graph neural network based weakly supervised learning framework to predict the methylation state of gene groups exhibiting coherent patterns across samples. Using data from three cohorts from The Cancer Genome Atlas (TCGA) - TCGA-LGG (Brain Lower Grade Glioma), TCGA-GBM (Glioblastoma Multiforme) ($n$=729) and TCGA-KIRC (Kidney Renal Clear Cell Carcinoma) ($n$=511) - we demonstrate that the proposed approach achieves significantly higher AUROC scores than the state-of-the-art (SOTA) methods, by more than $20\%$. We conduct gene set enrichment analyses on the gene groups and show that majority of the gene groups are significantly enriched in important hallmarks and pathways. We also generate spatially enriched heatmaps to further investigate links between histological patterns and DNA methylation states. To the best of our knowledge, this is the first study that explores association of spatially resolved histological patterns with gene group methylation states across multiple cancer types using weakly supervised deep learning.
From Traditional to Deep Learning Approaches in Whole Slide Image Registration: A Methodological Review
Feb 26, 2025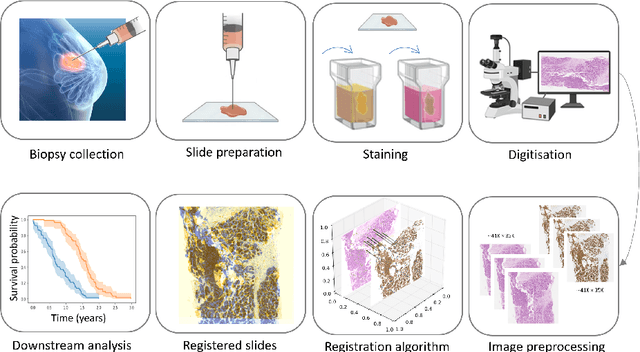

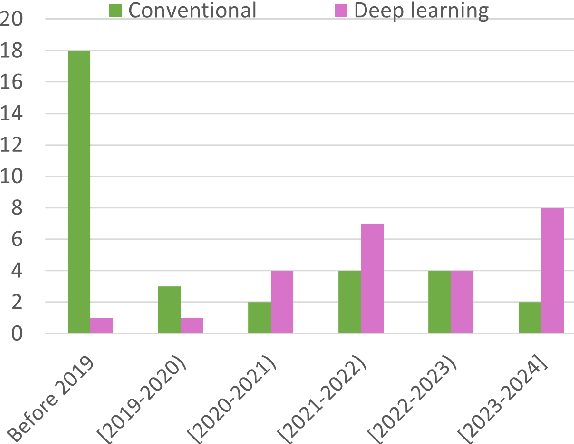
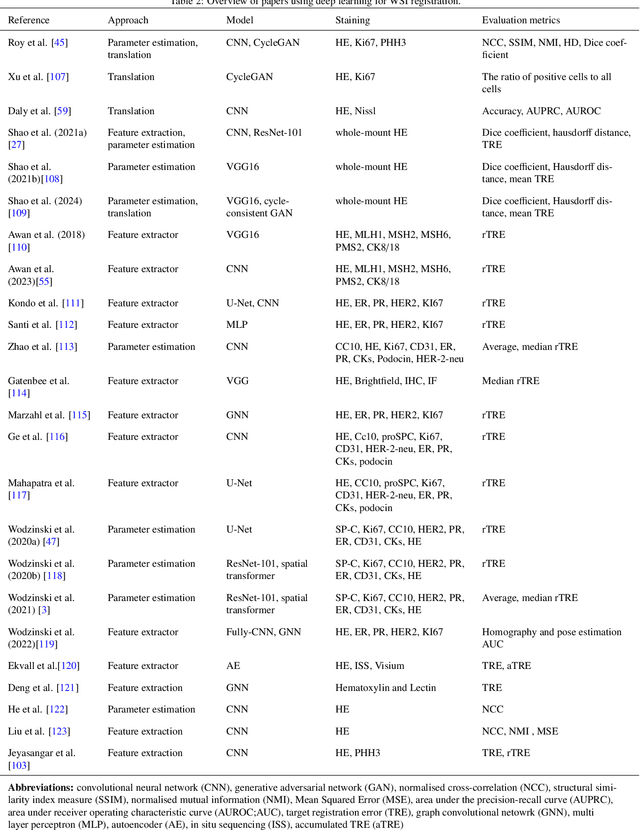
Whole slide image (WSI) registration is an essential task for analysing the tumour microenvironment (TME) in histopathology. It involves the alignment of spatial information between WSIs of the same section or serial sections of a tissue sample. The tissue sections are usually stained with single or multiple biomarkers before imaging, and the goal is to identify neighbouring nuclei along the Z-axis for creating a 3D image or identifying subclasses of cells in the TME. This task is considerably more challenging compared to radiology image registration, such as magnetic resonance imaging or computed tomography, due to various factors. These include gigapixel size of images, variations in appearance between differently stained tissues, changes in structure and morphology between non-consecutive sections, and the presence of artefacts, tears, and deformations. Currently, there is a noticeable gap in the literature regarding a review of the current approaches and their limitations, as well as the challenges and opportunities they present. We aim to provide a comprehensive understanding of the available approaches and their application for various purposes. Furthermore, we investigate current deep learning methods used for WSI registration, emphasising their diverse methodologies. We examine the available datasets and explore tools and software employed in the field. Finally, we identify open challenges and potential future trends in this area of research.
CellOMaps: A Compact Representation for Robust Classification of Lung Adenocarcinoma Growth Patterns
Jan 14, 2025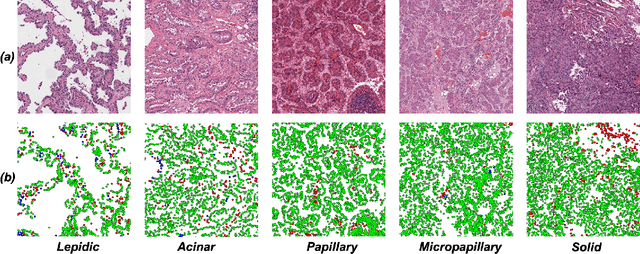



Lung adenocarcinoma (LUAD) is a morphologically heterogeneous disease, characterized by five primary histological growth patterns. The classification of such patterns is crucial due to their direct relation to prognosis but the high subjectivity and observer variability pose a major challenge. Although several studies have developed machine learning methods for growth pattern classification, they either only report the predominant pattern per slide or lack proper evaluation. We propose a generalizable machine learning pipeline capable of classifying lung tissue into one of the five patterns or as non-tumor. The proposed pipeline's strength lies in a novel compact Cell Organization Maps (cellOMaps) representation that captures the cellular spatial patterns from Hematoxylin and Eosin whole slide images (WSIs). The proposed pipeline provides state-of-the-art performance on LUAD growth pattern classification when evaluated on both internal unseen slides and external datasets, significantly outperforming the current approaches. In addition, our preliminary results show that the model's outputs can be used to predict patients Tumor Mutational Burden (TMB) levels.
CoNIC Challenge: Pushing the Frontiers of Nuclear Detection, Segmentation, Classification and Counting
Mar 14, 2023



Nuclear detection, segmentation and morphometric profiling are essential in helping us further understand the relationship between histology and patient outcome. To drive innovation in this area, we setup a community-wide challenge using the largest available dataset of its kind to assess nuclear segmentation and cellular composition. Our challenge, named CoNIC, stimulated the development of reproducible algorithms for cellular recognition with real-time result inspection on public leaderboards. We conducted an extensive post-challenge analysis based on the top-performing models using 1,658 whole-slide images of colon tissue. With around 700 million detected nuclei per model, associated features were used for dysplasia grading and survival analysis, where we demonstrated that the challenge's improvement over the previous state-of-the-art led to significant boosts in downstream performance. Our findings also suggest that eosinophils and neutrophils play an important role in the tumour microevironment. We release challenge models and WSI-level results to foster the development of further methods for biomarker discovery.
Mimicking a Pathologist: Dual Attention Model for Scoring of Gigapixel Histology Images
Feb 19, 2023


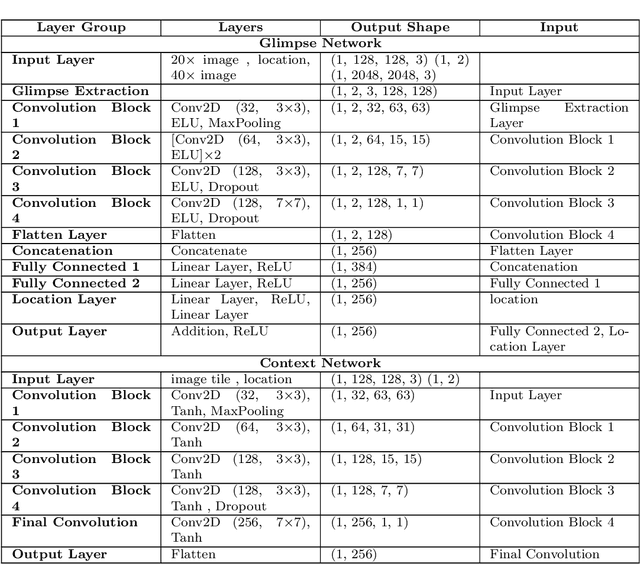
Some major challenges associated with the automated processing of whole slide images (WSIs) includes their sheer size, different magnification levels and high resolution. Utilizing these images directly in AI frameworks is computationally expensive due to memory constraints, while downsampling WSIs incurs information loss and splitting WSIs into tiles and patches results in loss of important contextual information. We propose a novel dual attention approach, consisting of two main components, to mimic visual examination by a pathologist. The first component is a soft attention model which takes as input a high-level view of the WSI to determine various regions of interest. We employ a custom sampling method to extract diverse and spatially distinct image tiles from selected high attention areas. The second component is a hard attention classification model, which further extracts a sequence of multi-resolution glimpses from each tile for classification. Since hard attention is non-differentiable, we train this component using reinforcement learning and predict the location of glimpses without processing all patches of a given tile, thereby aligning with pathologist's way of diagnosis. We train our components both separately and in an end-to-end fashion using a joint loss function to demonstrate the efficacy of our proposed model. We employ our proposed model on two different IHC use cases: HER2 prediction on breast cancer and prediction of Intact/Loss status of two MMR biomarkers, for colorectal cancer. We show that the proposed model achieves accuracy comparable to state-of-the-art methods while only processing a small fraction of the WSI at highest magnification.
Consistency Regularisation in Varying Contexts and Feature Perturbations for Semi-Supervised Semantic Segmentation of Histology Images
Feb 11, 2023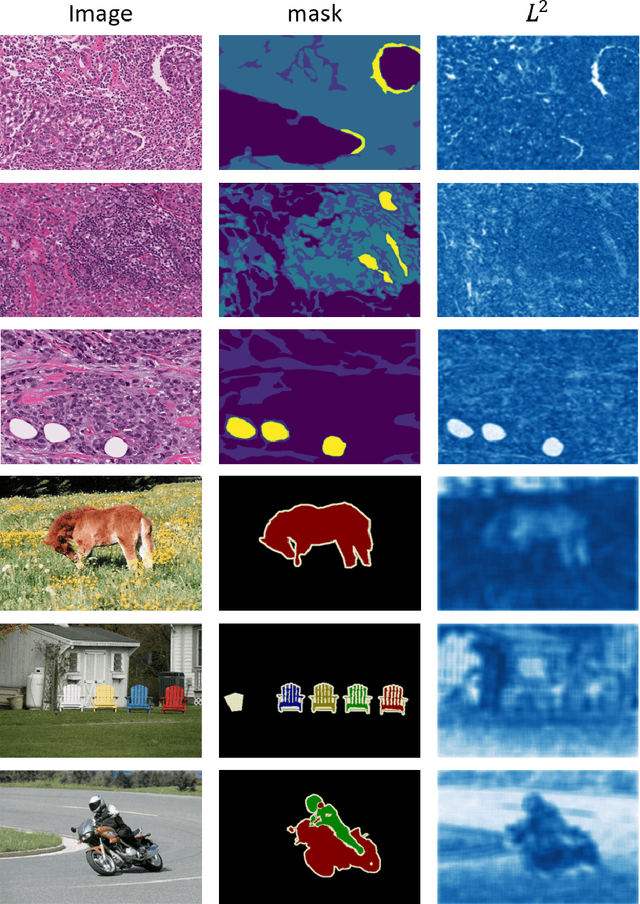
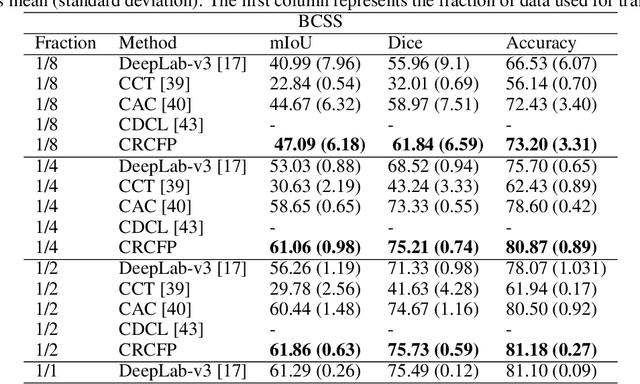
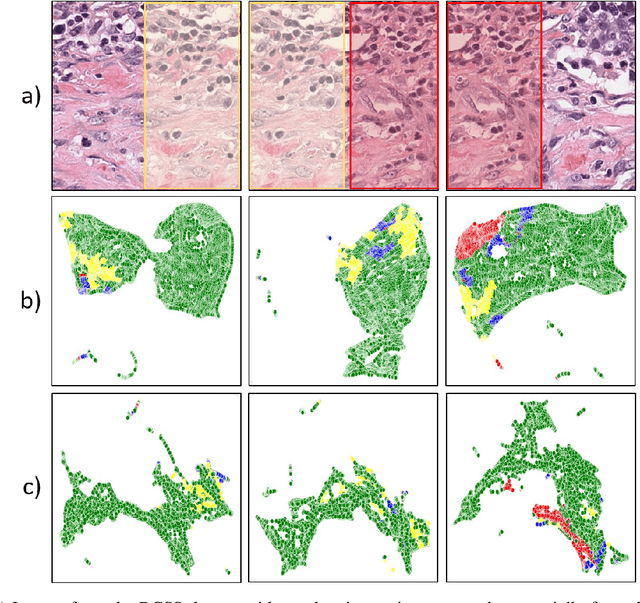
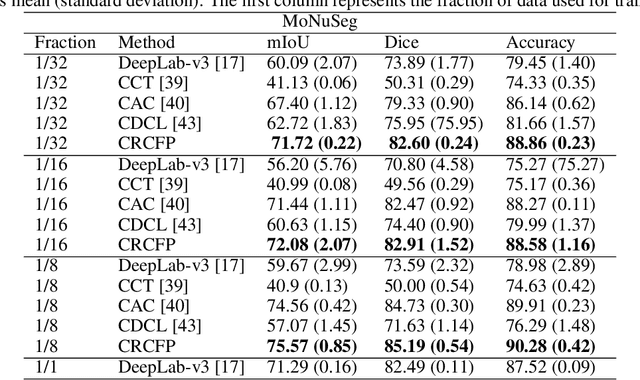
Semantic segmentation of various tissue and nuclei types in histology images is fundamental to many downstream tasks in the area of computational pathology (CPath). In recent years, Deep Learning (DL) methods have been shown to perform well on segmentation tasks but DL methods generally require a large amount of pixel-wise annotated data. Pixel-wise annotation sometimes requires expert's knowledge and time which is laborious and costly to obtain. In this paper, we present a consistency based semi-supervised learning (SSL) approach that can help mitigate this challenge by exploiting a large amount of unlabelled data for model training thus alleviating the need for a large annotated dataset. However, SSL models might also be susceptible to changing context and features perturbations exhibiting poor generalisation due to the limited training data. We propose an SSL method that learns robust features from both labelled and unlabelled images by enforcing consistency against varying contexts and feature perturbations. The proposed method incorporates context-aware consistency by contrasting pairs of overlapping images in a pixel-wise manner from changing contexts resulting in robust and context invariant features. We show that cross-consistency training makes the encoder features invariant to different perturbations and improves the prediction confidence. Finally, entropy minimisation is employed to further boost the confidence of the final prediction maps from unlabelled data. We conduct an extensive set of experiments on two publicly available large datasets (BCSS and MoNuSeg) and show superior performance compared to the state-of-the-art methods.
Deep Feature based Cross-slide Registration
Feb 27, 2022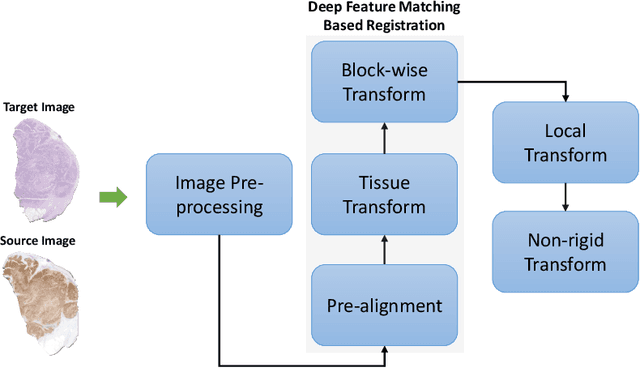
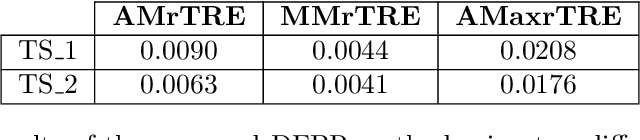
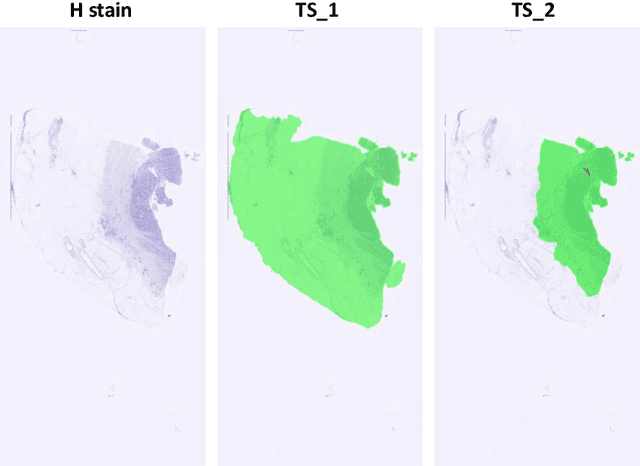
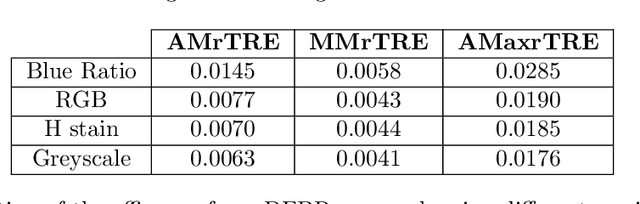
Cross-slide image analysis provides additional information by analysing the expression of different biomarkers as compared to a single slide analysis. Slides stained with different biomarkers are analysed side by side which may reveal unknown relations between the different biomarkers. During the slide preparation, a tissue section may be placed at an arbitrary orientation as compared to other sections of the same tissue block. The problem is compounded by the fact that tissue contents are likely to change from one section to the next and there may be unique artefacts on some of the slides. This makes registration of each section to a reference section of the same tissue block an important pre-requisite task before any cross-slide analysis. We propose a deep feature based registration (DFBR) method which utilises data-driven features to estimate the rigid transformation. We adopted a multi-stage strategy for improving the quality of registration. We also developed a visualisation tool to view registered pairs of WSIs at different magnifications. With the help of this tool, one can apply a transformation on the fly without the need to generate transformed source WSI in a pyramidal form. We compared the performance of data-driven features with that of hand-crafted features on the COMET dataset. Our approach can align the images with low registration errors. Generally, the success of non-rigid registration is dependent on the quality of rigid registration. To evaluate the efficacy of the DFBR method, the first two steps of the ANHIR winner's framework are replaced with our DFBR to register challenge provided image pairs. The modified framework produce comparable results to that of challenge winning team.
Deep Learning based Prediction of MSI in Colorectal Cancer via Prediction of the Status of MMR Markers
Feb 24, 2022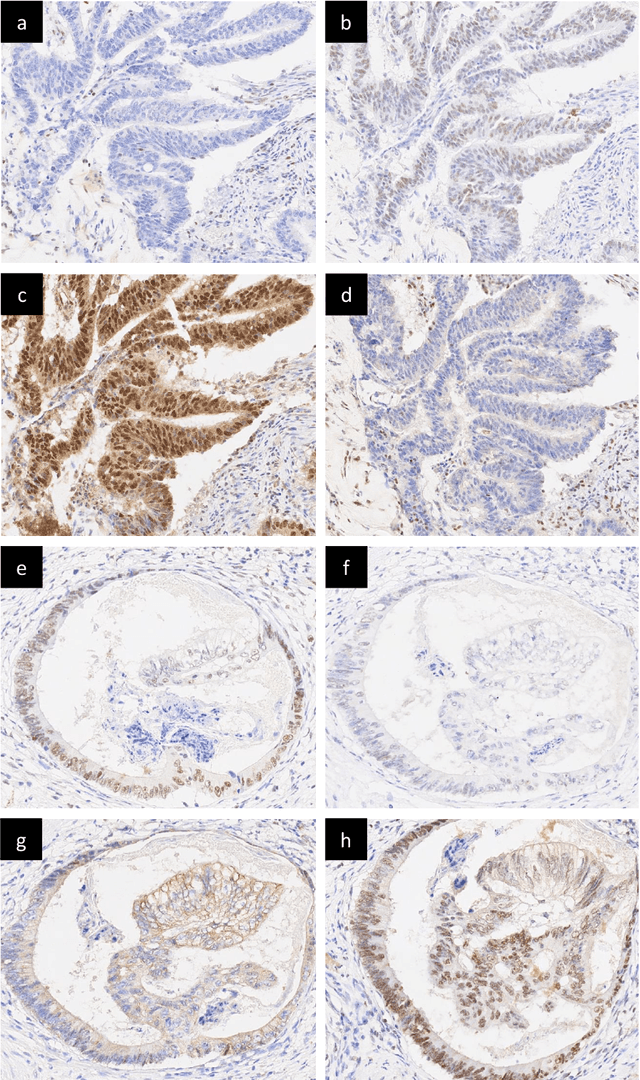

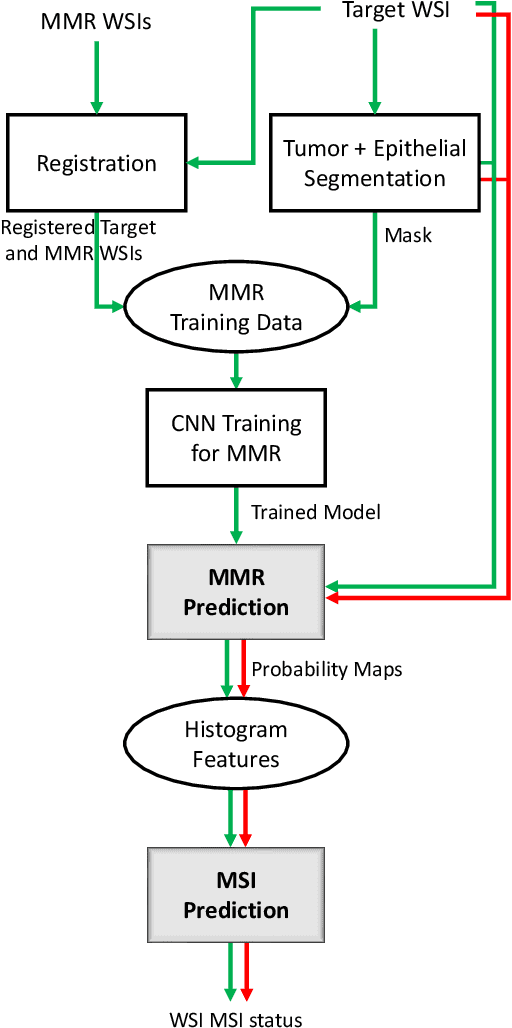
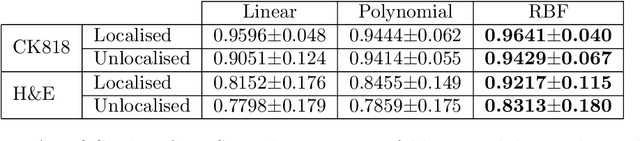
An accurate diagnosis and profiling of tumour are critical to the best treatment choices for cancer patients. In addition to the cancer type and its aggressiveness, molecular heterogeneity also plays a vital role in treatment selection. MSI or MMR deficiency is one of the well-studied aberrations in terms of molecular changes. Colorectal cancer patients with MMR deficiency respond well to immunotherapy, hence assessment of the relevant molecular markers can assist clinicians in making optimal treatment selections for patients. Immunohistochemistry is one of the ways for identifying these molecular changes which requires additional sections of tumour tissue. Introduction of automated methods that can predict MSI or MMR status from a target image without the need for additional sections can substantially reduce the cost associated with it. In this work, we present our work on predicting MSI status in a two-stage process using a single target slide either stained with CK818 or H\&E. First, we train a multi-headed convolutional neural network model where each head is responsible for predicting one of the MMR protein expressions. To this end, we perform registration of MMR slides to the target slide as a pre-processing step. In the second stage, statistical features computed from the MMR prediction maps are used for the final MSI prediction. Our results demonstrate that MSI classification can be improved on incorporating fine-grained MMR labels in comparison to the previous approaches in which coarse labels (MSI/MSS) are utilised.
FedDropoutAvg: Generalizable federated learning for histopathology image classification
Nov 25, 2021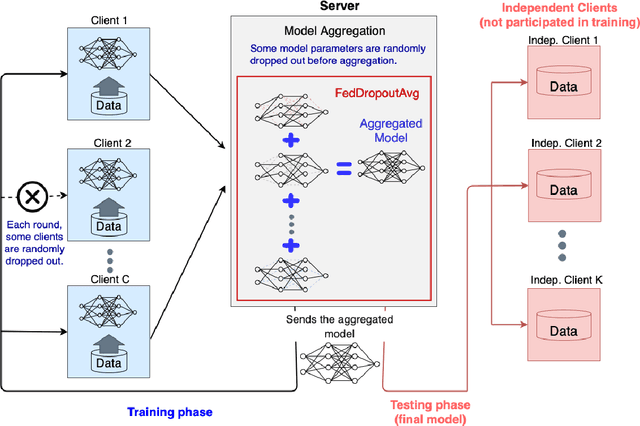
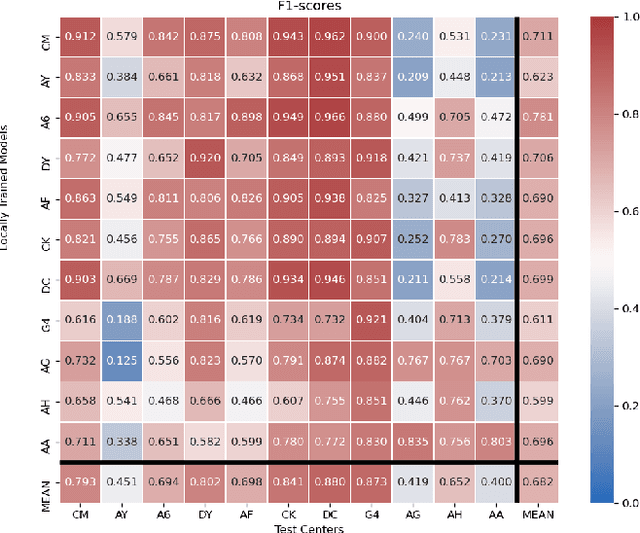
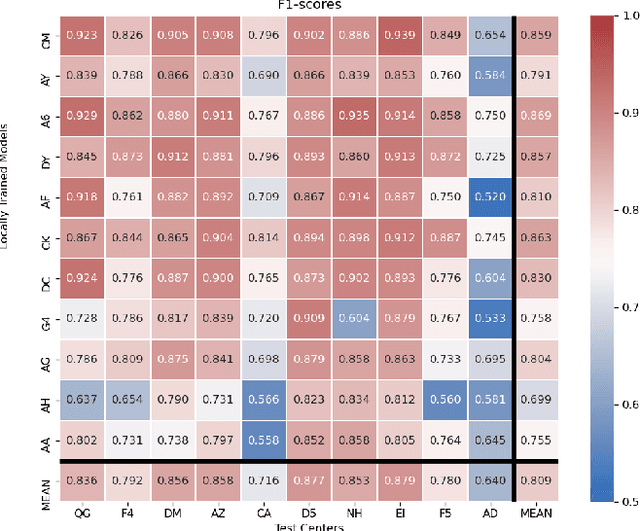
Federated learning (FL) enables collaborative learning of a deep learning model without sharing the data of participating sites. FL in medical image analysis tasks is relatively new and open for enhancements. In this study, we propose FedDropoutAvg, a new federated learning approach for training a generalizable model. The proposed method takes advantage of randomness, both in client selection and also in federated averaging process. We compare FedDropoutAvg to several algorithms in an FL scenario for real-world multi-site histopathology image classification task. We show that with FedDropoutAvg, the final model can achieve performance better than other FL approaches and closer to a classical deep learning model that requires all data to be shared for centralized training. We test the trained models on a large dataset consisting of 1.2 million image tiles from 21 different centers. To evaluate the generalization ability of the proposed approach, we use held-out test sets from centers whose data was used in the FL and for unseen data from other independent centers whose data was not used in the federated training. We show that the proposed approach is more generalizable than other state-of-the-art federated training approaches. To the best of our knowledge, ours is the first study to use a randomized client and local model parameter selection procedure in a federated setting for a medical image analysis task.
Simultaneous Nuclear Instance and Layer Segmentation in Oral Epithelial Dysplasia
Sep 01, 2021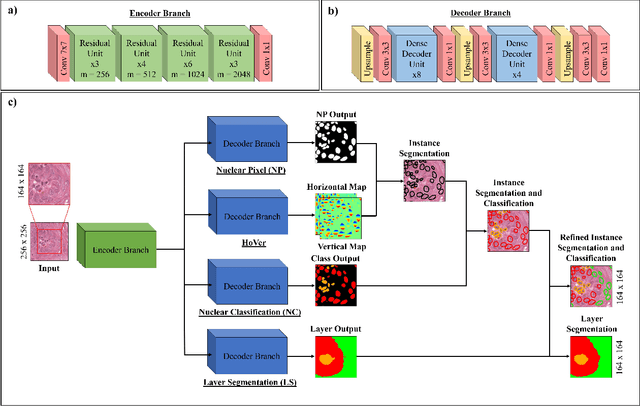
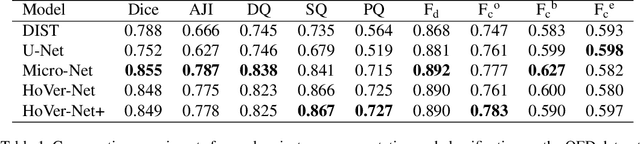
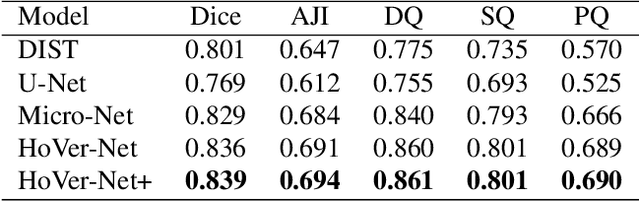
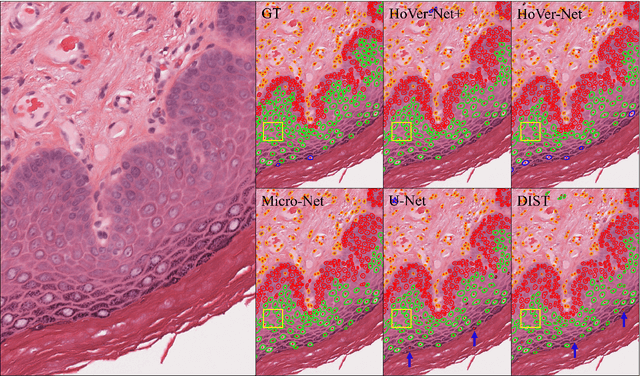
Oral epithelial dysplasia (OED) is a pre-malignant histopathological diagnosis given to lesions of the oral cavity. Predicting OED grade or whether a case will transition to malignancy is critical for early detection and appropriate treatment. OED typically begins in the lower third of the epithelium before progressing upwards with grade severity, thus we have suggested that segmenting intra-epithelial layers, in addition to individual nuclei, may enable researchers to evaluate important layer-specific morphological features for grade/malignancy prediction. We present HoVer-Net+, a deep learning framework to simultaneously segment (and classify) nuclei and (intra-)epithelial layers in H&E stained slides from OED cases. The proposed architecture consists of an encoder branch and four decoder branches for simultaneous instance segmentation of nuclei and semantic segmentation of the epithelial layers. We show that the proposed model achieves the state-of-the-art (SOTA) performance in both tasks, with no additional costs when compared to previous SOTA methods for each task. To the best of our knowledge, ours is the first method for simultaneous nuclear instance segmentation and semantic tissue segmentation, with potential for use in computational pathology for other similar simultaneous tasks and for future studies into malignancy prediction.