 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDeep Learning based Prediction of MSI in Colorectal Cancer via Prediction of the Status of MMR Markers
Feb 24, 2022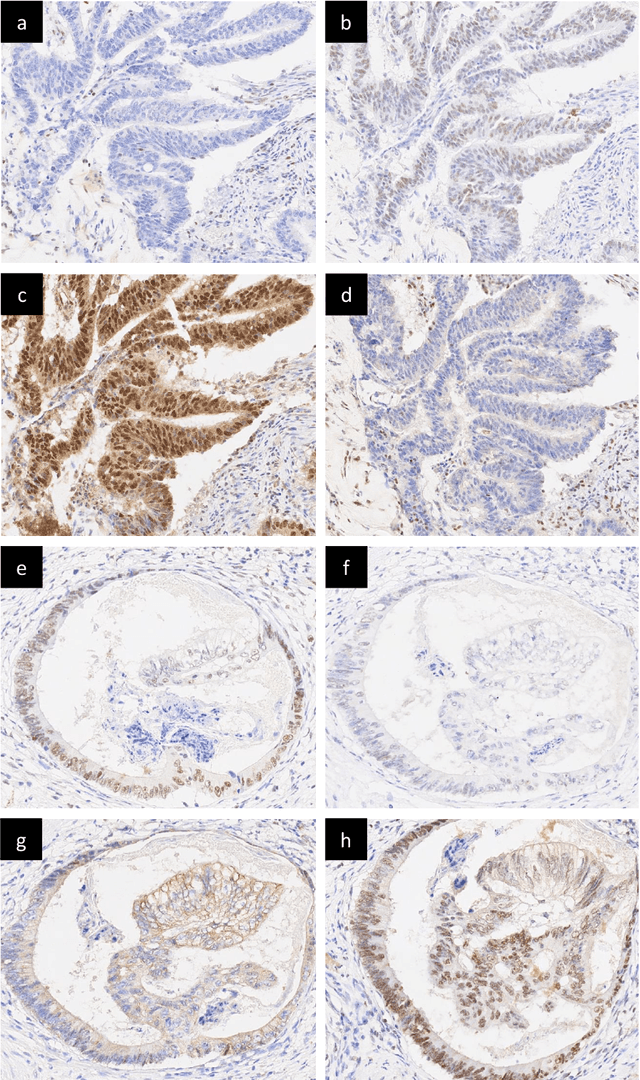

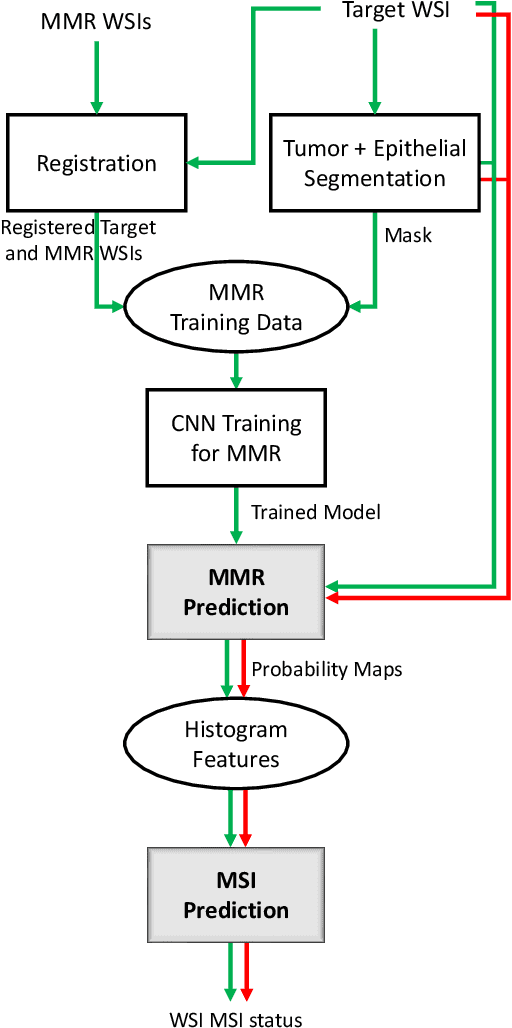
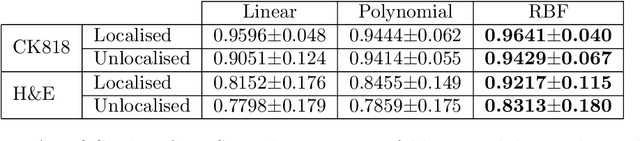
An accurate diagnosis and profiling of tumour are critical to the best treatment choices for cancer patients. In addition to the cancer type and its aggressiveness, molecular heterogeneity also plays a vital role in treatment selection. MSI or MMR deficiency is one of the well-studied aberrations in terms of molecular changes. Colorectal cancer patients with MMR deficiency respond well to immunotherapy, hence assessment of the relevant molecular markers can assist clinicians in making optimal treatment selections for patients. Immunohistochemistry is one of the ways for identifying these molecular changes which requires additional sections of tumour tissue. Introduction of automated methods that can predict MSI or MMR status from a target image without the need for additional sections can substantially reduce the cost associated with it. In this work, we present our work on predicting MSI status in a two-stage process using a single target slide either stained with CK818 or H\&E. First, we train a multi-headed convolutional neural network model where each head is responsible for predicting one of the MMR protein expressions. To this end, we perform registration of MMR slides to the target slide as a pre-processing step. In the second stage, statistical features computed from the MMR prediction maps are used for the final MSI prediction. Our results demonstrate that MSI classification can be improved on incorporating fine-grained MMR labels in comparison to the previous approaches in which coarse labels (MSI/MSS) are utilised.
Lizard: A Large-Scale Dataset for Colonic Nuclear Instance Segmentation and Classification
Aug 25, 2021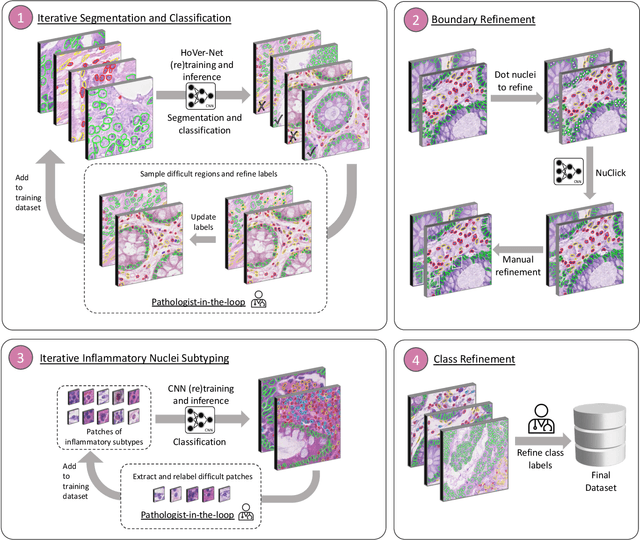
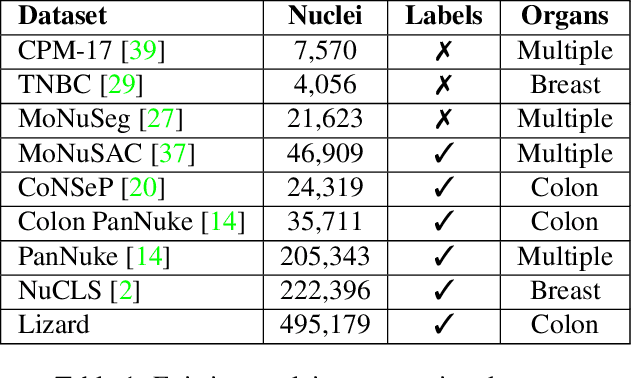
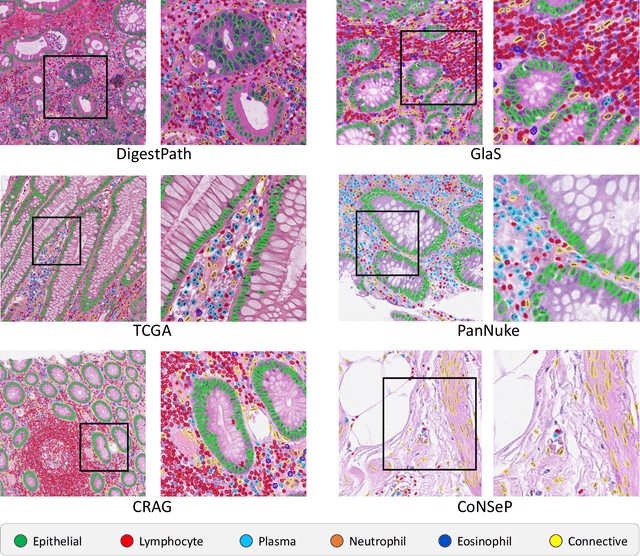
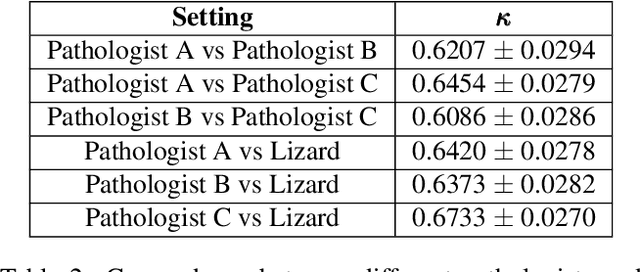
The development of deep segmentation models for computational pathology (CPath) can help foster the investigation of interpretable morphological biomarkers. Yet, there is a major bottleneck in the success of such approaches because supervised deep learning models require an abundance of accurately labelled data. This issue is exacerbated in the field of CPath because the generation of detailed annotations usually demands the input of a pathologist to be able to distinguish between different tissue constructs and nuclei. Manually labelling nuclei may not be a feasible approach for collecting large-scale annotated datasets, especially when a single image region can contain thousands of different cells. However, solely relying on automatic generation of annotations will limit the accuracy and reliability of ground truth. Therefore, to help overcome the above challenges, we propose a multi-stage annotation pipeline to enable the collection of large-scale datasets for histology image analysis, with pathologist-in-the-loop refinement steps. Using this pipeline, we generate the largest known nuclear instance segmentation and classification dataset, containing nearly half a million labelled nuclei in H&E stained colon tissue. We have released the dataset and encourage the research community to utilise it to drive forward the development of downstream cell-based models in CPath.