 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLizard: A Large-Scale Dataset for Colonic Nuclear Instance Segmentation and Classification
Aug 25, 2021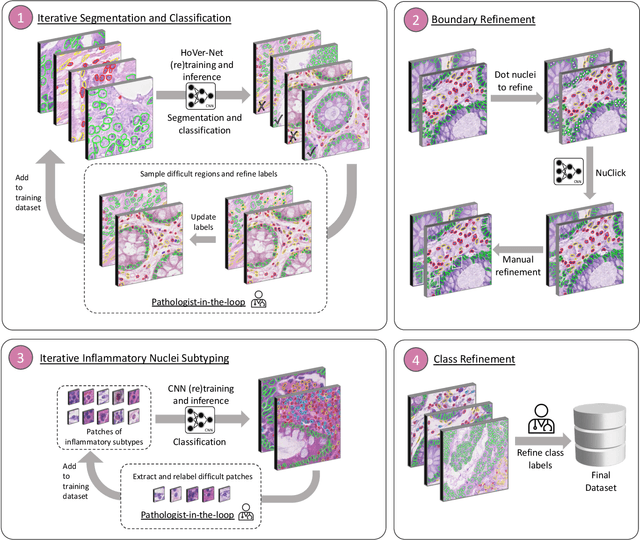
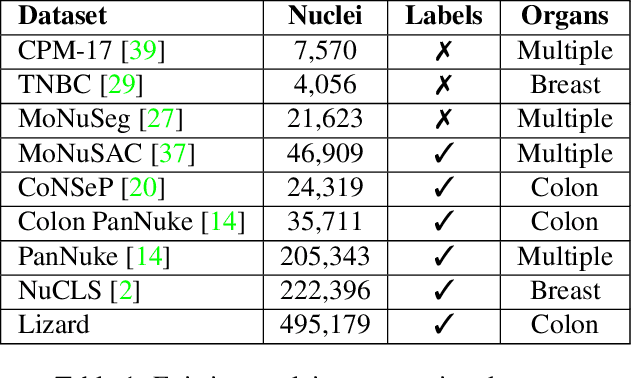
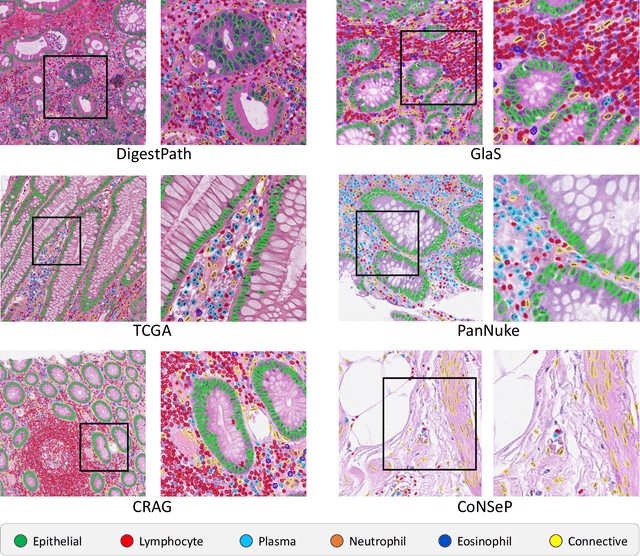
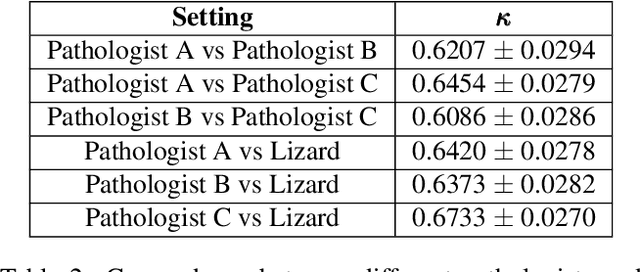
The development of deep segmentation models for computational pathology (CPath) can help foster the investigation of interpretable morphological biomarkers. Yet, there is a major bottleneck in the success of such approaches because supervised deep learning models require an abundance of accurately labelled data. This issue is exacerbated in the field of CPath because the generation of detailed annotations usually demands the input of a pathologist to be able to distinguish between different tissue constructs and nuclei. Manually labelling nuclei may not be a feasible approach for collecting large-scale annotated datasets, especially when a single image region can contain thousands of different cells. However, solely relying on automatic generation of annotations will limit the accuracy and reliability of ground truth. Therefore, to help overcome the above challenges, we propose a multi-stage annotation pipeline to enable the collection of large-scale datasets for histology image analysis, with pathologist-in-the-loop refinement steps. Using this pipeline, we generate the largest known nuclear instance segmentation and classification dataset, containing nearly half a million labelled nuclei in H&E stained colon tissue. We have released the dataset and encourage the research community to utilise it to drive forward the development of downstream cell-based models in CPath.
Fast and Accurate Tumor Segmentation of Histology Images using Persistent Homology and Deep Convolutional Features
May 09, 2018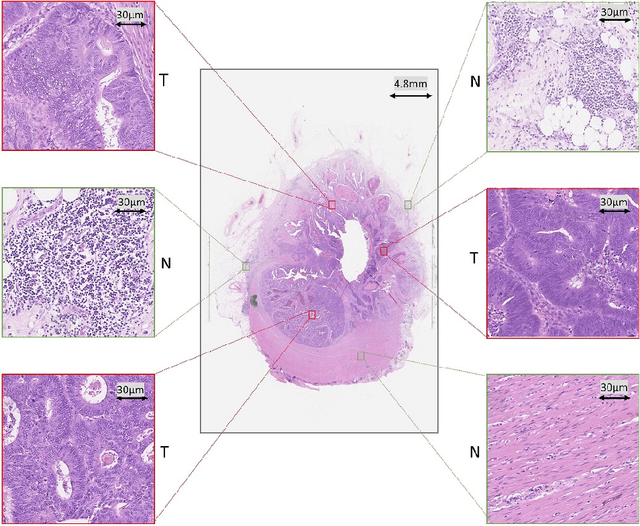
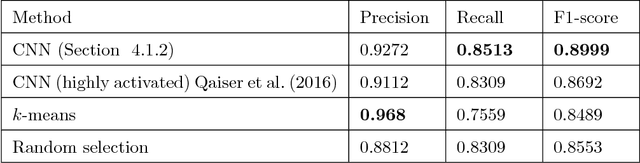

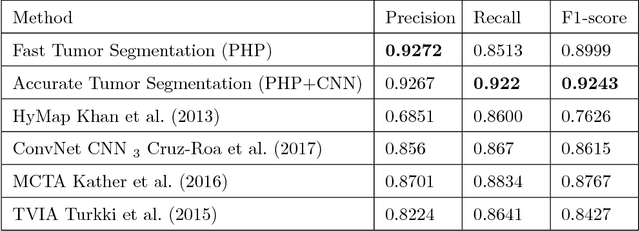
Tumor segmentation in whole-slide images of histology slides is an important step towards computer-assisted diagnosis. In this work, we propose a tumor segmentation framework based on the novel concept of persistent homology profiles (PHPs). For a given image patch, the homology profiles are derived by efficient computation of persistent homology, which is an algebraic tool from homology theory. We propose an efficient way of computing topological persistence of an image, alternative to simplicial homology. The PHPs are devised to distinguish tumor regions from their normal counterparts by modeling the atypical characteristics of tumor nuclei. We propose two variants of our method for tumor segmentation: one that targets speed without compromising accuracy and the other that targets higher accuracy. The fast version is based on the selection of exemplar image patches from a convolution neural network (CNN) and patch classification by quantifying the divergence between the PHPs of exemplars and the input image patch. Detailed comparative evaluation shows that the proposed algorithm is significantly faster than competing algorithms while achieving comparable results. The accurate version combines the PHPs and high-level CNN features and employs a multi-stage ensemble strategy for image patch labeling. Experimental results demonstrate that the combination of PHPs and CNN features outperforms competing algorithms. This study is performed on two independently collected colorectal datasets containing adenoma, adenocarcinoma, signet and healthy cases. Collectively, the accurate tumor segmentation produces the highest average patch-level F1-score, as compared with competing algorithms, on malignant and healthy cases from both the datasets. Overall the proposed framework highlights the utility of persistent homology for histopathology image analysis.