 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe case for delegated AI autonomy for Human AI teaming in healthcare
Mar 24, 2025In this paper we propose an advanced approach to integrating artificial intelligence (AI) into healthcare: autonomous decision support. This approach allows the AI algorithm to act autonomously for a subset of patient cases whilst serving a supportive role in other subsets of patient cases based on defined delegation criteria. By leveraging the complementary strengths of both humans and AI, it aims to deliver greater overall performance than existing human-AI teaming models. It ensures safe handling of patient cases and potentially reduces clinician review time, whilst being mindful of AI tool limitations. After setting the approach within the context of current human-AI teaming models, we outline the delegation criteria and apply them to a specific AI-based tool used in histopathology. The potential impact of the approach and the regulatory requirements for its successful implementation are then discussed.
An Automated Pipeline for Tumour-Infiltrating Lymphocyte Scoring in Breast Cancer
Nov 21, 2023Tumour-infiltrating lymphocytes (TILs) are considered as a valuable prognostic markers in both triple-negative and human epidermal growth factor receptor 2 (HER2) positive breast cancer. In this study, we introduce an innovative deep learning pipeline based on the Efficient-UNet architecture to predict the TILs score for breast cancer whole-slide images (WSIs). We first segment tumour and stromal regions in order to compute a tumour bulk mask. We then detect TILs within the tumour-associated stroma, generating a TILs score by closely mirroring the pathologist's workflow. Our method exhibits state-of-the-art performance in segmenting tumour/stroma areas and TILs detection, as demonstrated by internal cross-validation on the TiGER Challenge training dataset and evaluation on the final leaderboards. Additionally, our TILs score proves competitive in predicting survival outcomes within the same challenge, underscoring the clinical relevance and potential of our automated TILs scoring pipeline as a breast cancer prognostic tool.
CoNIC Challenge: Pushing the Frontiers of Nuclear Detection, Segmentation, Classification and Counting
Mar 14, 2023



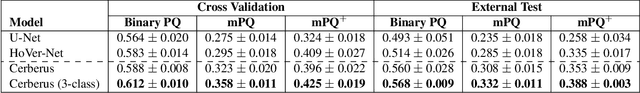
Nuclear detection, segmentation and morphometric profiling are essential in helping us further understand the relationship between histology and patient outcome. To drive innovation in this area, we setup a community-wide challenge using the largest available dataset of its kind to assess nuclear segmentation and cellular composition. Our challenge, named CoNIC, stimulated the development of reproducible algorithms for cellular recognition with real-time result inspection on public leaderboards. We conducted an extensive post-challenge analysis based on the top-performing models using 1,658 whole-slide images of colon tissue. With around 700 million detected nuclei per model, associated features were used for dysplasia grading and survival analysis, where we demonstrated that the challenge's improvement over the previous state-of-the-art led to significant boosts in downstream performance. Our findings also suggest that eosinophils and neutrophils play an important role in the tumour microevironment. We release challenge models and WSI-level results to foster the development of further methods for biomarker discovery.
LYSTO: The Lymphocyte Assessment Hackathon and Benchmark Dataset
Jan 16, 2023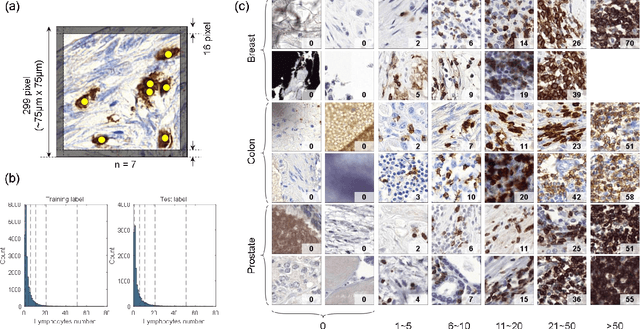
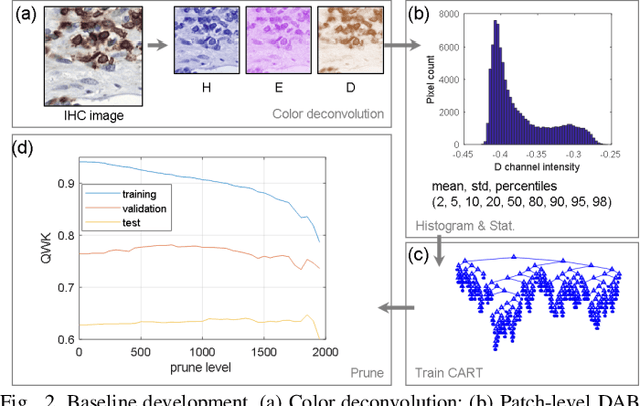
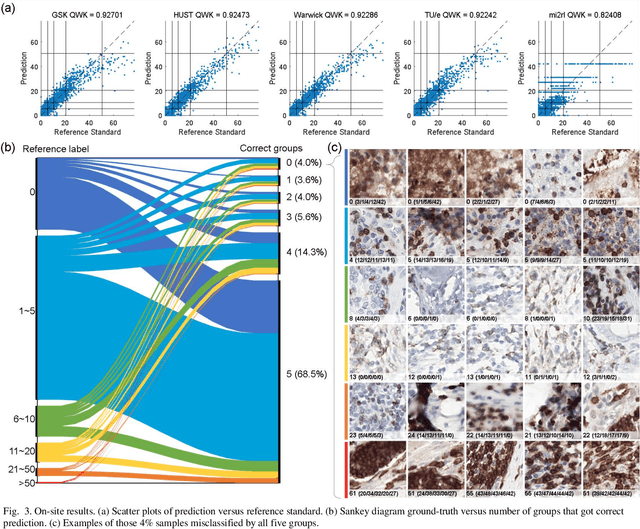
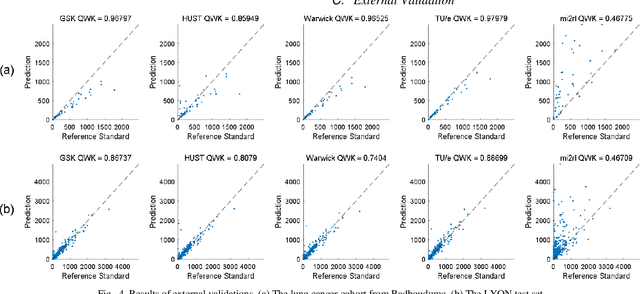
We introduce LYSTO, the Lymphocyte Assessment Hackathon, which was held in conjunction with the MICCAI 2019 Conference in Shenzen (China). The competition required participants to automatically assess the number of lymphocytes, in particular T-cells, in histopathological images of colon, breast, and prostate cancer stained with CD3 and CD8 immunohistochemistry. Differently from other challenges setup in medical image analysis, LYSTO participants were solely given a few hours to address this problem. In this paper, we describe the goal and the multi-phase organization of the hackathon; we describe the proposed methods and the on-site results. Additionally, we present post-competition results where we show how the presented methods perform on an independent set of lung cancer slides, which was not part of the initial competition, as well as a comparison on lymphocyte assessment between presented methods and a panel of pathologists. We show that some of the participants were capable to achieve pathologist-level performance at lymphocyte assessment. After the hackathon, LYSTO was left as a lightweight plug-and-play benchmark dataset on grand-challenge website, together with an automatic evaluation platform. LYSTO has supported a number of research in lymphocyte assessment in oncology. LYSTO will be a long-lasting educational challenge for deep learning and digital pathology, it is available at https://lysto.grand-challenge.org/.
Nuclear Segmentation and Classification: On Color & Compression Generalization
Jan 09, 2023Since the introduction of digital and computational pathology as a field, one of the major problems in the clinical application of algorithms has been the struggle to generalize well to examples outside the distribution of the training data. Existing work to address this in both pathology and natural images has focused almost exclusively on classification tasks. We explore and evaluate the robustness of the 7 best performing nuclear segmentation and classification models from the largest computational pathology challenge for this problem to date, the CoNIC challenge. We demonstrate that existing state-of-the-art (SoTA) models are robust towards compression artifacts but suffer substantial performance reduction when subjected to shifts in the color domain. We find that using stain normalization to address the domain shift problem can be detrimental to the model performance. On the other hand, neural style transfer is more consistent in improving test performance when presented with large color variations in the wild.
IMPaSh: A Novel Domain-shift Resistant Representation for Colorectal Cancer Tissue Classification
Aug 23, 2022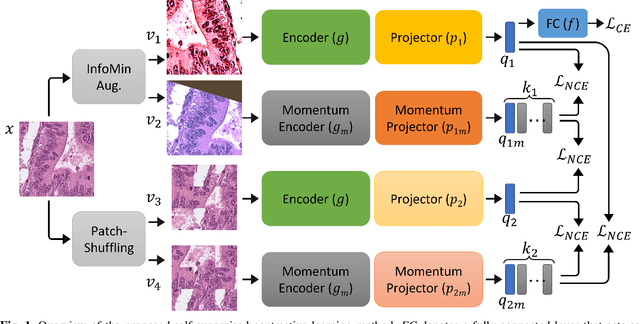
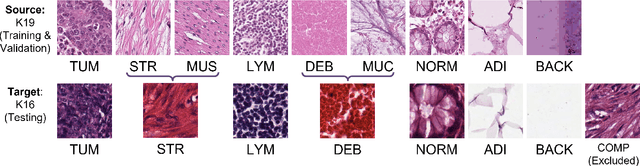
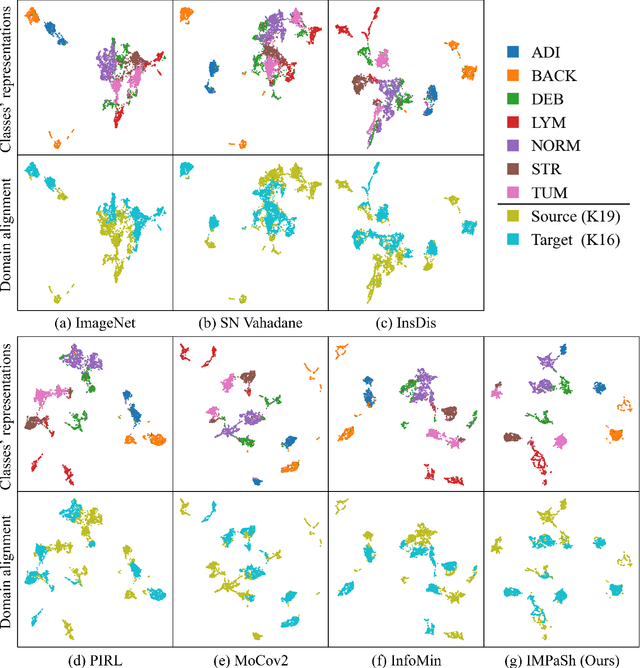
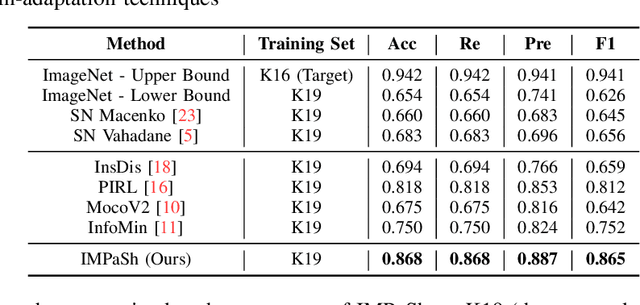
The appearance of histopathology images depends on tissue type, staining and digitization procedure. These vary from source to source and are the potential causes for domain-shift problems. Owing to this problem, despite the great success of deep learning models in computational pathology, a model trained on a specific domain may still perform sub-optimally when we apply them to another domain. To overcome this, we propose a new augmentation called PatchShuffling and a novel self-supervised contrastive learning framework named IMPaSh for pre-training deep learning models. Using these, we obtained a ResNet50 encoder that can extract image representation resistant to domain-shift. We compared our derived representation against those acquired based on other domain-generalization techniques by using them for the cross-domain classification of colorectal tissue images. We show that the proposed method outperforms other traditional histology domain-adaptation and state-of-the-art self-supervised learning methods. Code is available at: https://github.com/trinhvg/IMPash .
TIAger: Tumor-Infiltrating Lymphocyte Scoring in Breast Cancer for the TiGER Challenge
Jun 23, 2022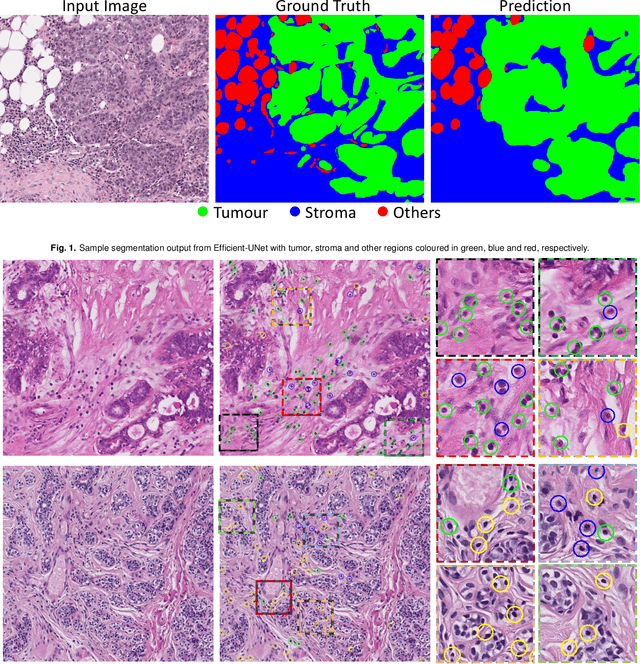
The quantification of tumor-infiltrating lymphocytes (TILs) has been shown to be an independent predictor for prognosis of breast cancer patients. Typically, pathologists give an estimate of the proportion of the stromal region that contains TILs to obtain a TILs score. The Tumor InfiltratinG lymphocytes in breast cancER (TiGER) challenge, aims to assess the prognostic significance of computer-generated TILs scores for predicting survival as part of a Cox proportional hazards model. For this challenge, as the TIAger team, we have developed an algorithm to first segment tumor vs. stroma, before localising the tumor bulk region for TILs detection. Finally, we use these outputs to generate a TILs score for each case. On preliminary testing, our approach achieved a tumor-stroma weighted Dice score of 0.791 and a FROC score of 0.572 for lymphocytic detection. For predicting survival, our model achieved a C-index of 0.719. These results achieved first place across the preliminary testing leaderboards of the TiGER challenge.
One Model is All You Need: Multi-Task Learning Enables Simultaneous Histology Image Segmentation and Classification
Feb 28, 2022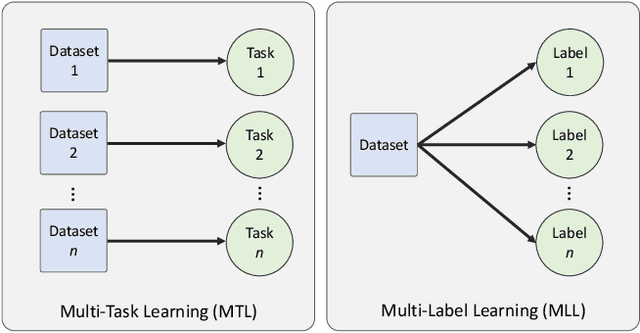
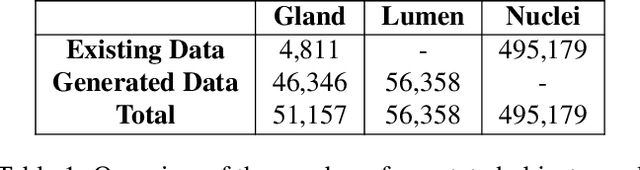
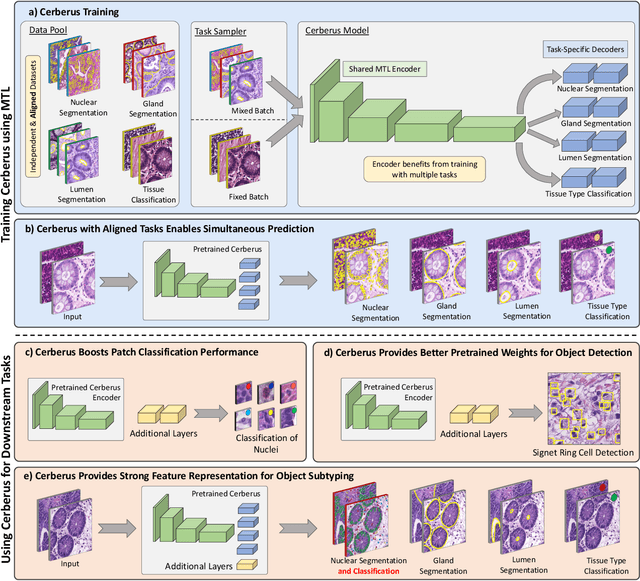
The recent surge in performance for image analysis of digitised pathology slides can largely be attributed to the advance of deep learning. Deep models can be used to initially localise various structures in the tissue and hence facilitate the extraction of interpretable features for biomarker discovery. However, these models are typically trained for a single task and therefore scale poorly as we wish to adapt the model for an increasing number of different tasks. Also, supervised deep learning models are very data hungry and therefore rely on large amounts of training data to perform well. In this paper we present a multi-task learning approach for segmentation and classification of nuclei, glands, lumen and different tissue regions that leverages data from multiple independent data sources. While ensuring that our tasks are aligned by the same tissue type and resolution, we enable simultaneous prediction with a single network. As a result of feature sharing, we also show that the learned representation can be used to improve downstream tasks, including nuclear classification and signet ring cell detection. As part of this work, we use a large dataset consisting of over 600K objects for segmentation and 440K patches for classification and make the data publicly available. We use our approach to process the colorectal subset of TCGA, consisting of 599 whole-slide images, to localise 377 million, 900K and 2.1 million nuclei, glands and lumen respectively. We make this resource available to remove a major barrier in the development of explainable models for computational pathology.
CoNIC: Colon Nuclei Identification and Counting Challenge 2022
Nov 29, 2021
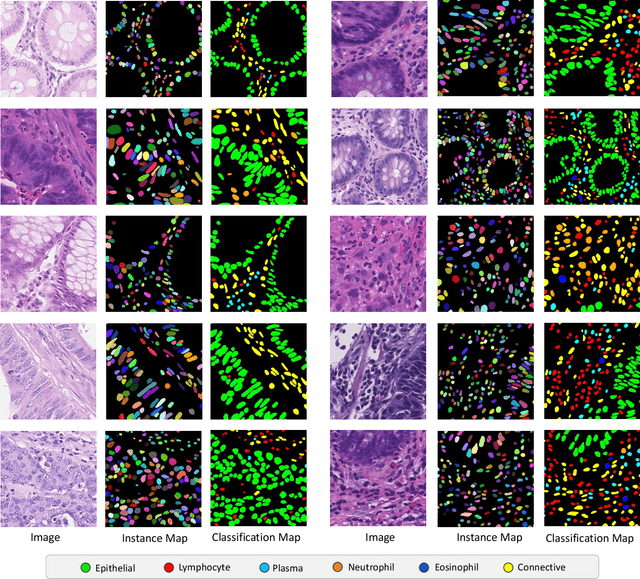
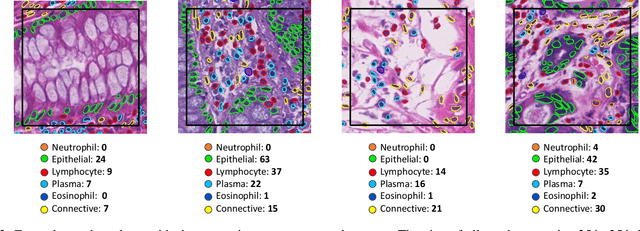
Nuclear segmentation, classification and quantification within Haematoxylin & Eosin stained histology images enables the extraction of interpretable cell-based features that can be used in downstream explainable models in computational pathology (CPath). However, automatic recognition of different nuclei is faced with a major challenge in that there are several different types of nuclei, some of them exhibiting large intra-class variability. To help drive forward research and innovation for automatic nuclei recognition in CPath, we organise the Colon Nuclei Identification and Counting (CoNIC) Challenge. The challenge encourages researchers to develop algorithms that perform segmentation, classification and counting of nuclei within the current largest known publicly available nuclei-level dataset in CPath, containing around half a million labelled nuclei. Therefore, the CoNIC challenge utilises over 10 times the number of nuclei as the previous largest challenge dataset for nuclei recognition. It is important for algorithms to be robust to input variation if we wish to deploy them in a clinical setting. Therefore, as part of this challenge we will also test the sensitivity of each submitted algorithm to certain input variations.
Simultaneous Nuclear Instance and Layer Segmentation in Oral Epithelial Dysplasia
Sep 01, 2021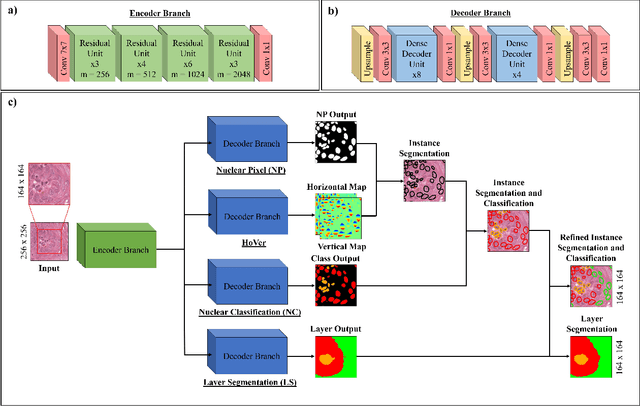
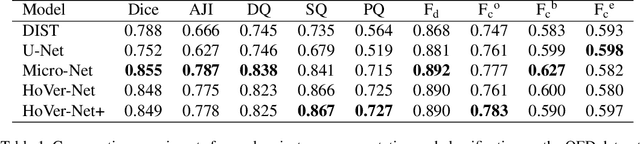
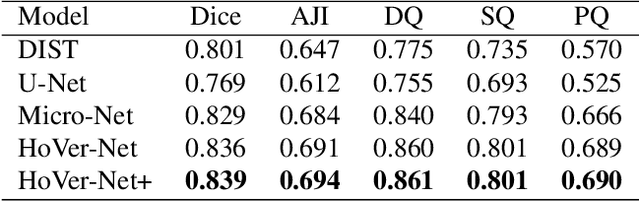
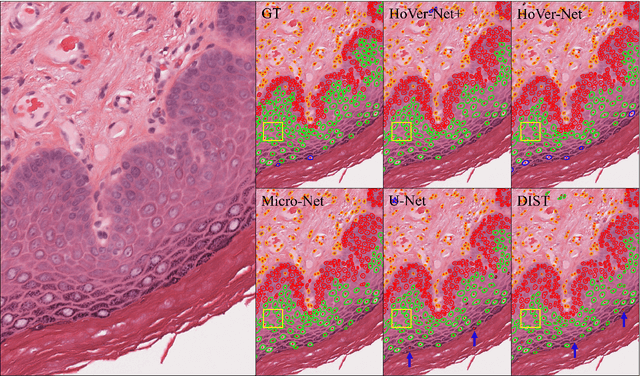
Oral epithelial dysplasia (OED) is a pre-malignant histopathological diagnosis given to lesions of the oral cavity. Predicting OED grade or whether a case will transition to malignancy is critical for early detection and appropriate treatment. OED typically begins in the lower third of the epithelium before progressing upwards with grade severity, thus we have suggested that segmenting intra-epithelial layers, in addition to individual nuclei, may enable researchers to evaluate important layer-specific morphological features for grade/malignancy prediction. We present HoVer-Net+, a deep learning framework to simultaneously segment (and classify) nuclei and (intra-)epithelial layers in H&E stained slides from OED cases. The proposed architecture consists of an encoder branch and four decoder branches for simultaneous instance segmentation of nuclei and semantic segmentation of the epithelial layers. We show that the proposed model achieves the state-of-the-art (SOTA) performance in both tasks, with no additional costs when compared to previous SOTA methods for each task. To the best of our knowledge, ours is the first method for simultaneous nuclear instance segmentation and semantic tissue segmentation, with potential for use in computational pathology for other similar simultaneous tasks and for future studies into malignancy prediction.