 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Novel Approach to Linking Histology Images with DNA Methylation
Apr 07, 2025DNA methylation is an epigenetic mechanism that regulates gene expression by adding methyl groups to DNA. Abnormal methylation patterns can disrupt gene expression and have been linked to cancer development. To quantify DNA methylation, specialized assays are typically used. However, these assays are often costly and have lengthy processing times, which limits their widespread availability in routine clinical practice. In contrast, whole slide images (WSIs) for the majority of cancer patients can be more readily available. As such, given the ready availability of WSIs, there is a compelling need to explore the potential relationship between WSIs and DNA methylation patterns. To address this, we propose an end-to-end graph neural network based weakly supervised learning framework to predict the methylation state of gene groups exhibiting coherent patterns across samples. Using data from three cohorts from The Cancer Genome Atlas (TCGA) - TCGA-LGG (Brain Lower Grade Glioma), TCGA-GBM (Glioblastoma Multiforme) ($n$=729) and TCGA-KIRC (Kidney Renal Clear Cell Carcinoma) ($n$=511) - we demonstrate that the proposed approach achieves significantly higher AUROC scores than the state-of-the-art (SOTA) methods, by more than $20\%$. We conduct gene set enrichment analyses on the gene groups and show that majority of the gene groups are significantly enriched in important hallmarks and pathways. We also generate spatially enriched heatmaps to further investigate links between histological patterns and DNA methylation states. To the best of our knowledge, this is the first study that explores association of spatially resolved histological patterns with gene group methylation states across multiple cancer types using weakly supervised deep learning.
HistoKernel: Whole Slide Image Level Maximum Mean Discrepancy Kernels for Pan-Cancer Predictive Modelling
Aug 09, 2024



Machine learning in computational pathology (CPath) often aggregates patch-level predictions from multi-gigapixel Whole Slide Images (WSIs) to generate WSI-level prediction scores for crucial tasks such as survival prediction and drug effect prediction. However, current methods do not explicitly characterize distributional differences between patch sets within WSIs. We introduce HistoKernel, a novel Maximum Mean Discrepancy (MMD) kernel that measures distributional similarity between WSIs for enhanced prediction performance on downstream prediction tasks. Our comprehensive analysis demonstrates HistoKernel's effectiveness across various machine learning tasks, including retrieval (n = 9,362), drug sensitivity regression (n = 551), point mutation classification (n = 3,419), and survival analysis (n = 2,291), outperforming existing deep learning methods. Additionally, HistoKernel seamlessly integrates multi-modal data and offers a novel perturbation-based method for patch-level explainability. This work pioneers the use of kernel-based methods for WSI-level predictive modeling, opening new avenues for research. Code is available at https://github.com/pkeller00/HistoKernel.
An Automated Pipeline for Tumour-Infiltrating Lymphocyte Scoring in Breast Cancer
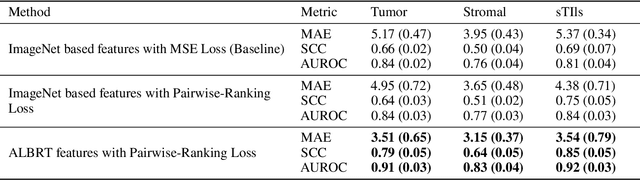
Nov 21, 2023Tumour-infiltrating lymphocytes (TILs) are considered as a valuable prognostic markers in both triple-negative and human epidermal growth factor receptor 2 (HER2) positive breast cancer. In this study, we introduce an innovative deep learning pipeline based on the Efficient-UNet architecture to predict the TILs score for breast cancer whole-slide images (WSIs). We first segment tumour and stromal regions in order to compute a tumour bulk mask. We then detect TILs within the tumour-associated stroma, generating a TILs score by closely mirroring the pathologist's workflow. Our method exhibits state-of-the-art performance in segmenting tumour/stroma areas and TILs detection, as demonstrated by internal cross-validation on the TiGER Challenge training dataset and evaluation on the final leaderboards. Additionally, our TILs score proves competitive in predicting survival outcomes within the same challenge, underscoring the clinical relevance and potential of our automated TILs scoring pipeline as a breast cancer prognostic tool.
CoNIC Challenge: Pushing the Frontiers of Nuclear Detection, Segmentation, Classification and Counting
Mar 14, 2023



Nuclear detection, segmentation and morphometric profiling are essential in helping us further understand the relationship between histology and patient outcome. To drive innovation in this area, we setup a community-wide challenge using the largest available dataset of its kind to assess nuclear segmentation and cellular composition. Our challenge, named CoNIC, stimulated the development of reproducible algorithms for cellular recognition with real-time result inspection on public leaderboards. We conducted an extensive post-challenge analysis based on the top-performing models using 1,658 whole-slide images of colon tissue. With around 700 million detected nuclei per model, associated features were used for dysplasia grading and survival analysis, where we demonstrated that the challenge's improvement over the previous state-of-the-art led to significant boosts in downstream performance. Our findings also suggest that eosinophils and neutrophils play an important role in the tumour microevironment. We release challenge models and WSI-level results to foster the development of further methods for biomarker discovery.
Maximum Mean Discrepancy Kernels for Predictive and Prognostic Modeling of Whole Slide Images
Jan 23, 2023How similar are two images? In computational pathology, where Whole Slide Images (WSIs) of digitally scanned tissue samples from patients can be multi-gigapixels in size, determination of degree of similarity between two WSIs is a challenging task with a number of practical applications. In this work, we explore a novel strategy based on kernelized Maximum Mean Discrepancy (MMD) analysis for determination of pairwise similarity between WSIs. The proposed approach works by calculating MMD between two WSIs using kernels over deep features of image patches. This allows representation of an entire dataset of WSIs as a kernel matrix for WSI level clustering, weakly-supervised prediction of TP-53 mutation status in breast cancer patients from their routine WSIs as well as survival analysis with state of the art prediction performance. We believe that this work will open up further avenues for application of WSI-level kernels for predictive and prognostic tasks in computational pathology.
SynCLay: Interactive Synthesis of Histology Images from Bespoke Cellular Layouts
Dec 28, 2022



Automated synthesis of histology images has several potential applications in computational pathology. However, no existing method can generate realistic tissue images with a bespoke cellular layout or user-defined histology parameters. In this work, we propose a novel framework called SynCLay (Synthesis from Cellular Layouts) that can construct realistic and high-quality histology images from user-defined cellular layouts along with annotated cellular boundaries. Tissue image generation based on bespoke cellular layouts through the proposed framework allows users to generate different histological patterns from arbitrary topological arrangement of different types of cells. SynCLay generated synthetic images can be helpful in studying the role of different types of cells present in the tumor microenvironmet. Additionally, they can assist in balancing the distribution of cellular counts in tissue images for designing accurate cellular composition predictors by minimizing the effects of data imbalance. We train SynCLay in an adversarial manner and integrate a nuclear segmentation and classification model in its training to refine nuclear structures and generate nuclear masks in conjunction with synthetic images. During inference, we combine the model with another parametric model for generating colon images and associated cellular counts as annotations given the grade of differentiation and cell densities of different cells. We assess the generated images quantitatively and report on feedback from trained pathologists who assigned realism scores to a set of images generated by the framework. The average realism score across all pathologists for synthetic images was as high as that for the real images. We also show that augmenting limited real data with the synthetic data generated by our framework can significantly boost prediction performance of the cellular composition prediction task.
TIAger: Tumor-Infiltrating Lymphocyte Scoring in Breast Cancer for the TiGER Challenge
Jun 23, 2022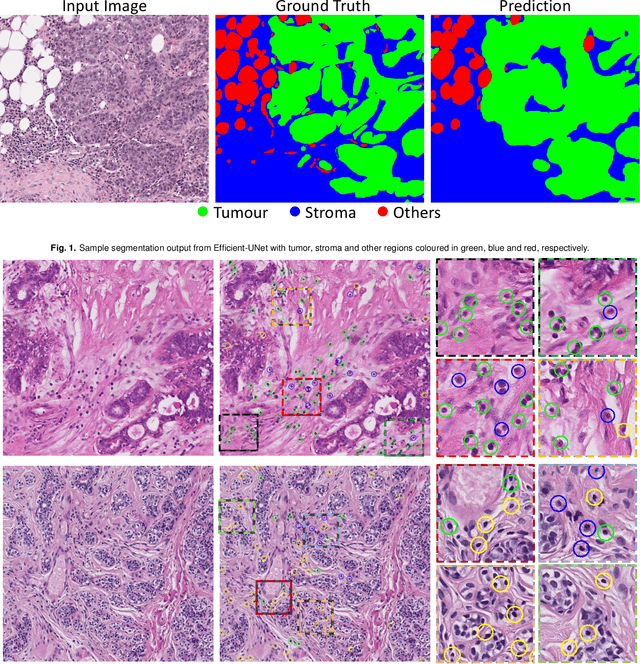
The quantification of tumor-infiltrating lymphocytes (TILs) has been shown to be an independent predictor for prognosis of breast cancer patients. Typically, pathologists give an estimate of the proportion of the stromal region that contains TILs to obtain a TILs score. The Tumor InfiltratinG lymphocytes in breast cancER (TiGER) challenge, aims to assess the prognostic significance of computer-generated TILs scores for predicting survival as part of a Cox proportional hazards model. For this challenge, as the TIAger team, we have developed an algorithm to first segment tumor vs. stroma, before localising the tumor bulk region for TILs detection. Finally, we use these outputs to generate a TILs score for each case. On preliminary testing, our approach achieved a tumor-stroma weighted Dice score of 0.791 and a FROC score of 0.572 for lymphocytic detection. For predicting survival, our model achieved a C-index of 0.719. These results achieved first place across the preliminary testing leaderboards of the TiGER challenge.
Cellular Segmentation and Composition in Routine Histology Images using Deep Learning
Mar 04, 2022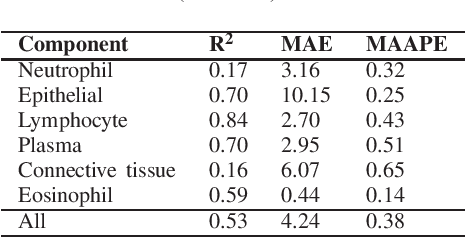
Identification and quantification of nuclei in colorectal cancer haematoxylin \& eosin (H\&E) stained histology images is crucial to prognosis and patient management. In computational pathology these tasks are referred to as nuclear segmentation, classification and composition and are used to extract meaningful interpretable cytological and architectural features for downstream analysis. The CoNIC challenge poses the task of automated nuclei segmentation, classification and composition into six different types of nuclei from the largest publicly known nuclei dataset - Lizard. In this regard, we have developed pipelines for the prediction of nuclei segmentation using HoVer-Net and ALBRT for cellular composition. On testing on the preliminary test set, HoVer-Net achieved a PQ of 0.58, a PQ+ of 0.58 and finally a mPQ+ of 0.35. For the prediction of cellular composition with ALBRT on the preliminary test set, we achieved an overall $R^2$ score of 0.53, consisting of 0.84 for lymphocytes, 0.70 for epithelial cells, 0.70 for plasma and .060 for eosinophils.
ALBRT: Cellular Composition Prediction in Routine Histology Images
Aug 26, 2021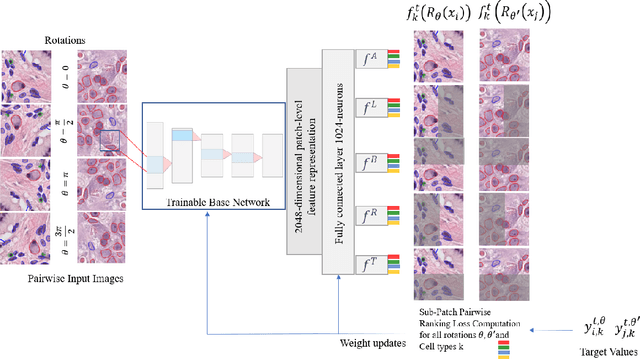
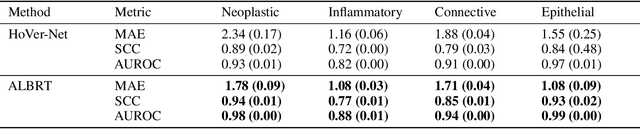
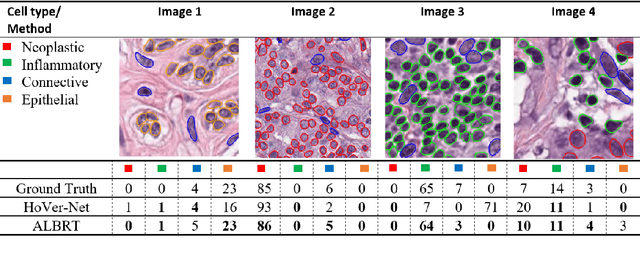
Cellular composition prediction, i.e., predicting the presence and counts of different types of cells in the tumor microenvironment from a digitized image of a Hematoxylin and Eosin (H&E) stained tissue section can be used for various tasks in computational pathology such as the analysis of cellular topology and interactions, subtype prediction, survival analysis, etc. In this work, we propose an image-based cellular composition predictor (ALBRT) which can accurately predict the presence and counts of different types of cells in a given image patch. ALBRT, by its contrastive-learning inspired design, learns a compact and rotation-invariant feature representation that is then used for cellular composition prediction of different cell types. It offers significant improvement over existing state-of-the-art approaches for cell classification and counting. The patch-level feature representation learned by ALBRT is transferrable for cellular composition analysis over novel datasets and can also be utilized for downstream prediction tasks in CPath as well. The code and the inference webserver for the proposed method are available at the URL: https://github.com/engrodawood/ALBRT.
All You Need is Color: Image based Spatial Gene Expression Prediction using Neural Stain Learning
Aug 26, 2021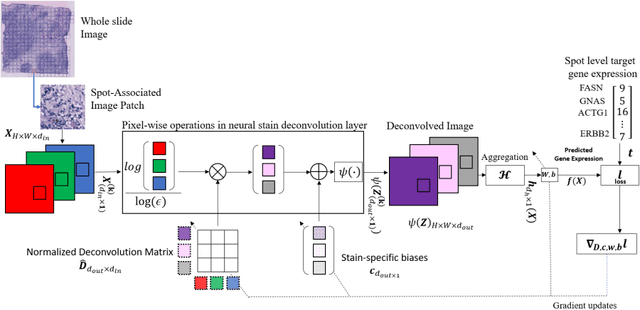
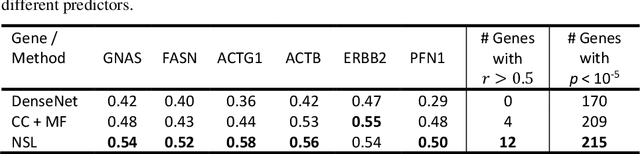
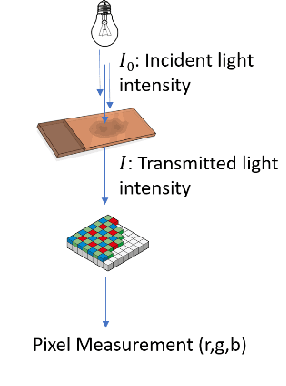
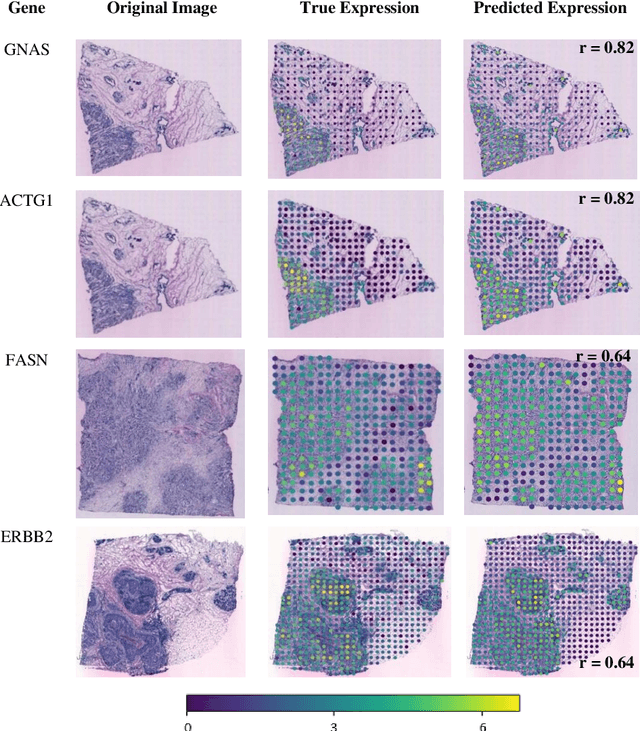
"Is it possible to predict expression levels of different genes at a given spatial location in the routine histology image of a tumor section by modeling its stain absorption characteristics?" In this work, we propose a "stain-aware" machine learning approach for prediction of spatial transcriptomic gene expression profiles using digital pathology image of a routine Hematoxylin & Eosin (H&E) histology section. Unlike recent deep learning methods which are used for gene expression prediction, our proposed approach termed Neural Stain Learning (NSL) explicitly models the association of stain absorption characteristics of the tissue with gene expression patterns in spatial transcriptomics by learning a problem-specific stain deconvolution matrix in an end-to-end manner. The proposed method with only 11 trainable weight parameters outperforms both classical regression models with cellular composition and morphological features as well as deep learning methods. We have found that the gene expression predictions from the proposed approach show higher correlations with true expression values obtained through sequencing for a larger set of genes in comparison to other approaches.