 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIn-silico biological discovery with large perturbation models
Mar 30, 2025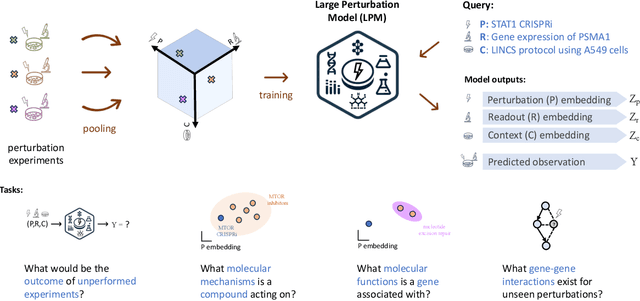
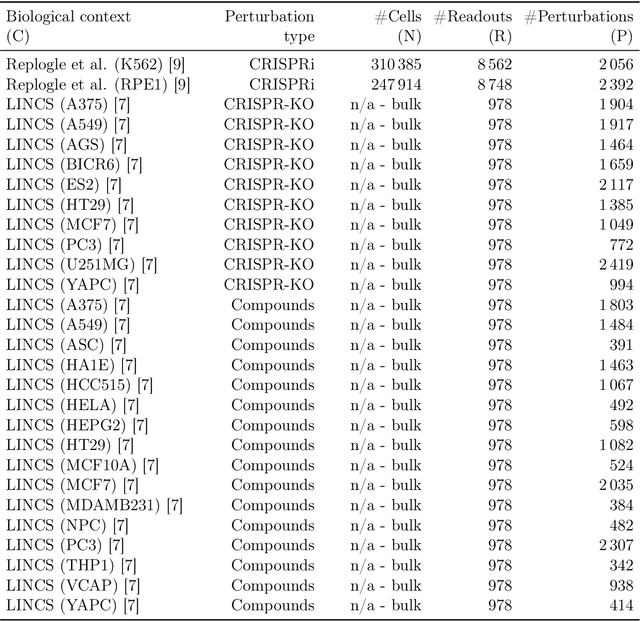
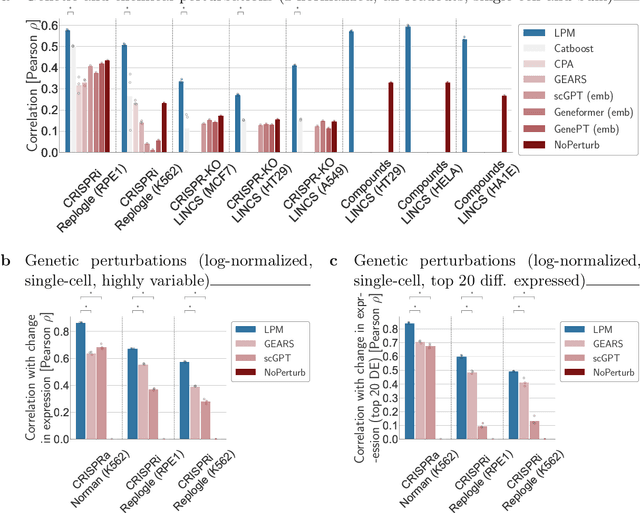
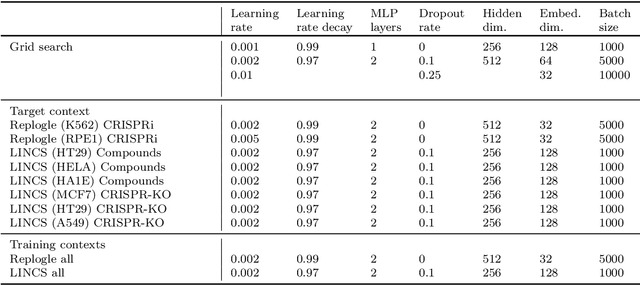
Data generated in perturbation experiments link perturbations to the changes they elicit and therefore contain information relevant to numerous biological discovery tasks -- from understanding the relationships between biological entities to developing therapeutics. However, these data encompass diverse perturbations and readouts, and the complex dependence of experimental outcomes on their biological context makes it challenging to integrate insights across experiments. Here, we present the Large Perturbation Model (LPM), a deep-learning model that integrates multiple, heterogeneous perturbation experiments by representing perturbation, readout, and context as disentangled dimensions. LPM outperforms existing methods across multiple biological discovery tasks, including in predicting post-perturbation transcriptomes of unseen experiments, identifying shared molecular mechanisms of action between chemical and genetic perturbations, and facilitating the inference of gene-gene interaction networks.
Multi-megabase scale genome interpretation with genetic language models
Jan 13, 2025Understanding how molecular changes caused by genetic variation drive disease risk is crucial for deciphering disease mechanisms. However, interpreting genome sequences is challenging because of the vast size of the human genome, and because its consequences manifest across a wide range of cells, tissues and scales -- spanning from molecular to whole organism level. Here, we present Phenformer, a multi-scale genetic language model that learns to generate mechanistic hypotheses as to how differences in genome sequence lead to disease-relevant changes in expression across cell types and tissues directly from DNA sequences of up to 88 million base pairs. Using whole genome sequencing data from more than 150 000 individuals, we show that Phenformer generates mechanistic hypotheses about disease-relevant cell and tissue types that match literature better than existing state-of-the-art methods, while using only sequence data. Furthermore, disease risk predictors enriched by Phenformer show improved prediction performance and generalisation to diverse populations. Accurate multi-megabase scale interpretation of whole genomes without additional experimental data enables both a deeper understanding of molecular mechanisms involved in disease and improved disease risk prediction at the level of individuals.