 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProtriever: End-to-End Differentiable Protein Homology Search for Fitness Prediction
Jun 10, 2025Retrieving homologous protein sequences is essential for a broad range of protein modeling tasks such as fitness prediction, protein design, structure modeling, and protein-protein interactions. Traditional workflows have relied on a two-step process: first retrieving homologs via Multiple Sequence Alignments (MSA), then training models on one or more of these alignments. However, MSA-based retrieval is computationally expensive, struggles with highly divergent sequences or complex insertions & deletions patterns, and operates independently of the downstream modeling objective. We introduce Protriever, an end-to-end differentiable framework that learns to retrieve relevant homologs while simultaneously training for the target task. When applied to protein fitness prediction, Protriever achieves state-of-the-art performance compared to sequence-based models that rely on MSA-based homolog retrieval, while being two orders of magnitude faster through efficient vector search. Protriever is both architecture- and task-agnostic, and can flexibly adapt to different retrieval strategies and protein databases at inference time -- offering a scalable alternative to alignment-centric approaches.
Multi-megabase scale genome interpretation with genetic language models
Jan 13, 2025Understanding how molecular changes caused by genetic variation drive disease risk is crucial for deciphering disease mechanisms. However, interpreting genome sequences is challenging because of the vast size of the human genome, and because its consequences manifest across a wide range of cells, tissues and scales -- spanning from molecular to whole organism level. Here, we present Phenformer, a multi-scale genetic language model that learns to generate mechanistic hypotheses as to how differences in genome sequence lead to disease-relevant changes in expression across cell types and tissues directly from DNA sequences of up to 88 million base pairs. Using whole genome sequencing data from more than 150 000 individuals, we show that Phenformer generates mechanistic hypotheses about disease-relevant cell and tissue types that match literature better than existing state-of-the-art methods, while using only sequence data. Furthermore, disease risk predictors enriched by Phenformer show improved prediction performance and generalisation to diverse populations. Accurate multi-megabase scale interpretation of whole genomes without additional experimental data enables both a deeper understanding of molecular mechanisms involved in disease and improved disease risk prediction at the level of individuals.
DiscoBAX: Discovery of Optimal Intervention Sets in Genomic Experiment Design
Dec 07, 2023The discovery of therapeutics to treat genetically-driven pathologies relies on identifying genes involved in the underlying disease mechanisms. Existing approaches search over the billions of potential interventions to maximize the expected influence on the target phenotype. However, to reduce the risk of failure in future stages of trials, practical experiment design aims to find a set of interventions that maximally change a target phenotype via diverse mechanisms. We propose DiscoBAX, a sample-efficient method for maximizing the rate of significant discoveries per experiment while simultaneously probing for a wide range of diverse mechanisms during a genomic experiment campaign. We provide theoretical guarantees of approximate optimality under standard assumptions, and conduct a comprehensive experimental evaluation covering both synthetic as well as real-world experimental design tasks. DiscoBAX outperforms existing state-of-the-art methods for experimental design, selecting effective and diverse perturbations in biological systems.
The CausalBench challenge: A machine learning contest for gene network inference from single-cell perturbation data
Aug 29, 2023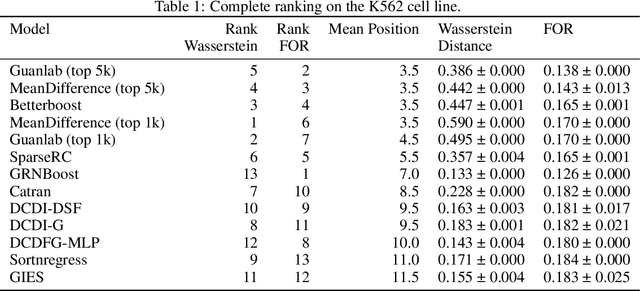
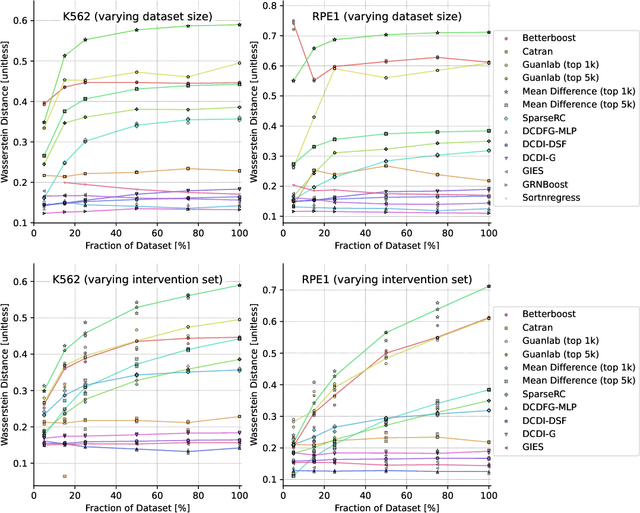
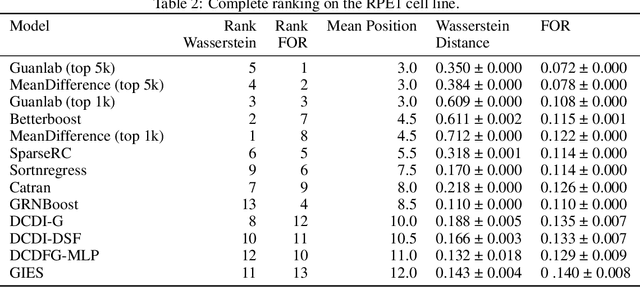
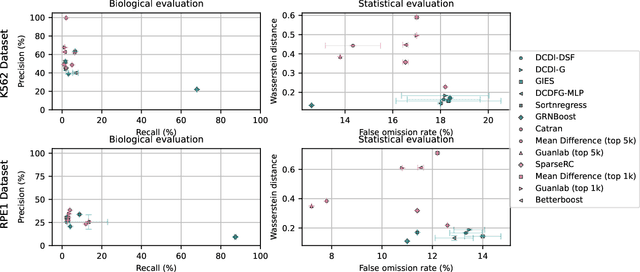
In drug discovery, mapping interactions between genes within cellular systems is a crucial early step. This helps formulate hypotheses regarding molecular mechanisms that could potentially be targeted by future medicines. The CausalBench Challenge was an initiative to invite the machine learning community to advance the state of the art in constructing gene-gene interaction networks. These networks, derived from large-scale, real-world datasets of single cells under various perturbations, are crucial for understanding the causal mechanisms underlying disease biology. Using the framework provided by the CausalBench benchmark, participants were tasked with enhancing the capacity of the state of the art methods to leverage large-scale genetic perturbation data. This report provides an analysis and summary of the methods submitted during the challenge to give a partial image of the state of the art at the time of the challenge. The winning solutions significantly improved performance compared to previous baselines, establishing a new state of the art for this critical task in biology and medicine.
Tranception: protein fitness prediction with autoregressive transformers and inference-time retrieval
May 27, 2022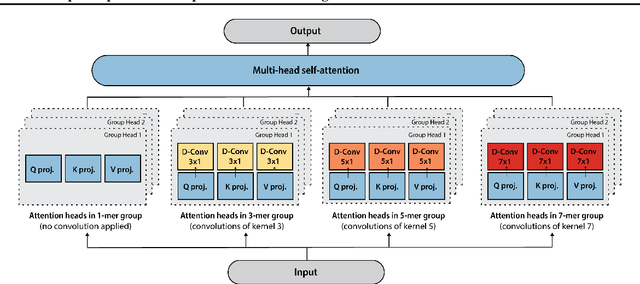
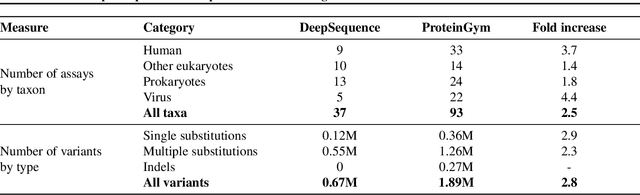
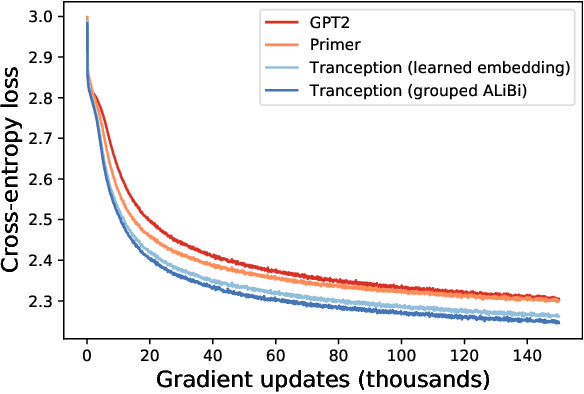
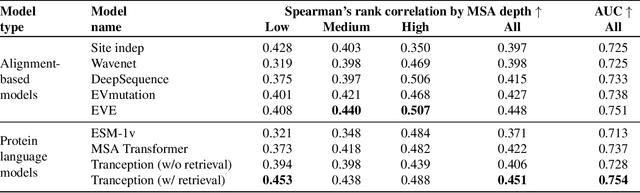
The ability to accurately model the fitness landscape of protein sequences is critical to a wide range of applications, from quantifying the effects of human variants on disease likelihood, to predicting immune-escape mutations in viruses and designing novel biotherapeutic proteins. Deep generative models of protein sequences trained on multiple sequence alignments have been the most successful approaches so far to address these tasks. The performance of these methods is however contingent on the availability of sufficiently deep and diverse alignments for reliable training. Their potential scope is thus limited by the fact many protein families are hard, if not impossible, to align. Large language models trained on massive quantities of non-aligned protein sequences from diverse families address these problems and show potential to eventually bridge the performance gap. We introduce Tranception, a novel transformer architecture leveraging autoregressive predictions and retrieval of homologous sequences at inference to achieve state-of-the-art fitness prediction performance. Given its markedly higher performance on multiple mutants, robustness to shallow alignments and ability to score indels, our approach offers significant gain of scope over existing approaches. To enable more rigorous model testing across a broader range of protein families, we develop ProteinGym -- an extensive set of multiplexed assays of variant effects, substantially increasing both the number and diversity of assays compared to existing benchmarks.
RITA: a Study on Scaling Up Generative Protein Sequence Models
May 11, 2022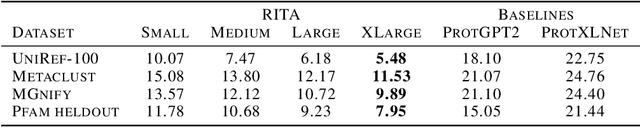
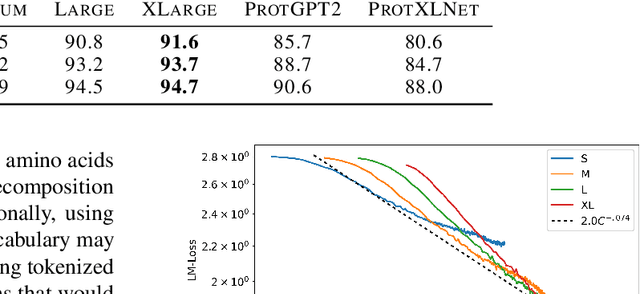
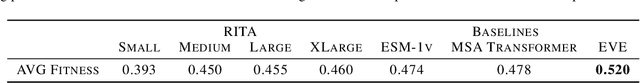
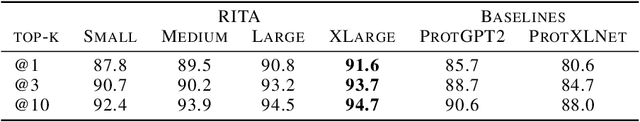
In this work we introduce RITA: a suite of autoregressive generative models for protein sequences, with up to 1.2 billion parameters, trained on over 280 million protein sequences belonging to the UniRef-100 database. Such generative models hold the promise of greatly accelerating protein design. We conduct the first systematic study of how capabilities evolve with model size for autoregressive transformers in the protein domain: we evaluate RITA models in next amino acid prediction, zero-shot fitness, and enzyme function prediction, showing benefits from increased scale. We release the RITA models openly, to the benefit of the research community.
GeneDisco: A Benchmark for Experimental Design in Drug Discovery
Oct 22, 2021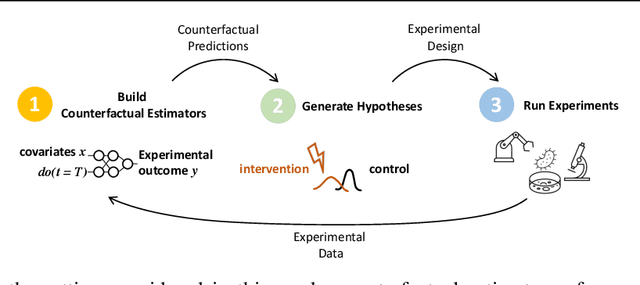
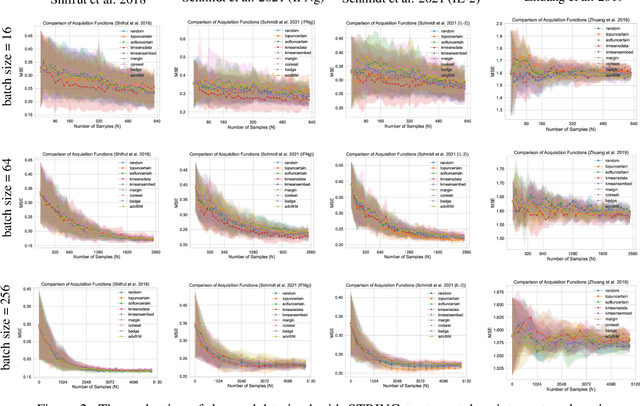
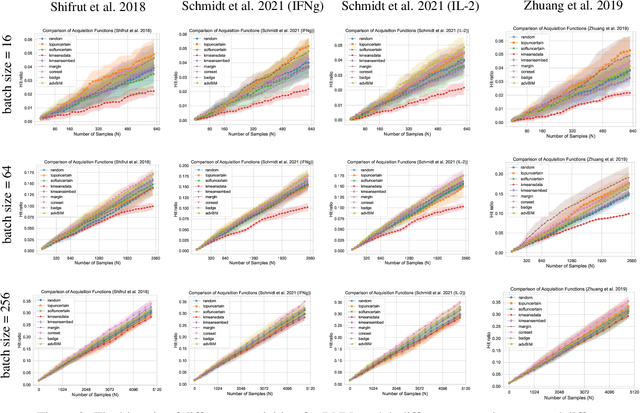
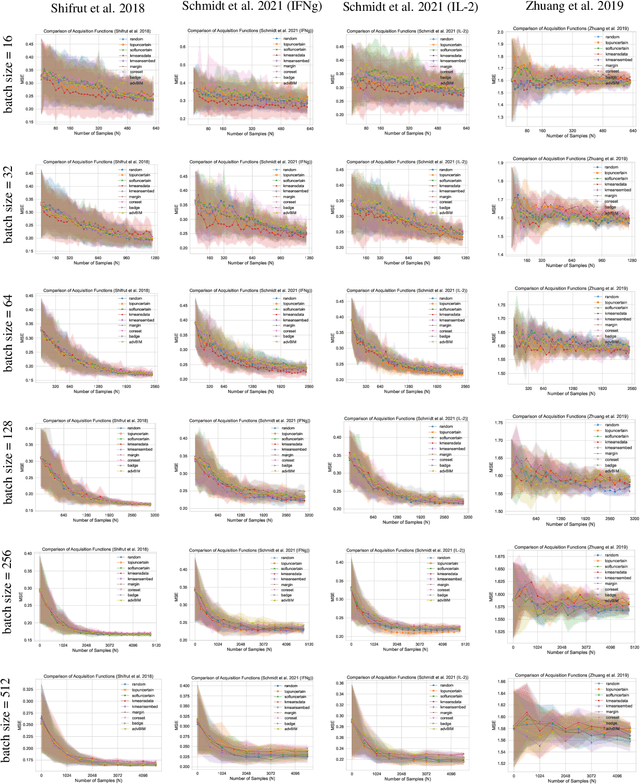
In vitro cellular experimentation with genetic interventions, using for example CRISPR technologies, is an essential step in early-stage drug discovery and target validation that serves to assess initial hypotheses about causal associations between biological mechanisms and disease pathologies. With billions of potential hypotheses to test, the experimental design space for in vitro genetic experiments is extremely vast, and the available experimental capacity - even at the largest research institutions in the world - pales in relation to the size of this biological hypothesis space. Machine learning methods, such as active and reinforcement learning, could aid in optimally exploring the vast biological space by integrating prior knowledge from various information sources as well as extrapolating to yet unexplored areas of the experimental design space based on available data. However, there exist no standardised benchmarks and data sets for this challenging task and little research has been conducted in this area to date. Here, we introduce GeneDisco, a benchmark suite for evaluating active learning algorithms for experimental design in drug discovery. GeneDisco contains a curated set of multiple publicly available experimental data sets as well as open-source implementations of state-of-the-art active learning policies for experimental design and exploration.
Improving black-box optimization in VAE latent space using decoder uncertainty
Jun 30, 2021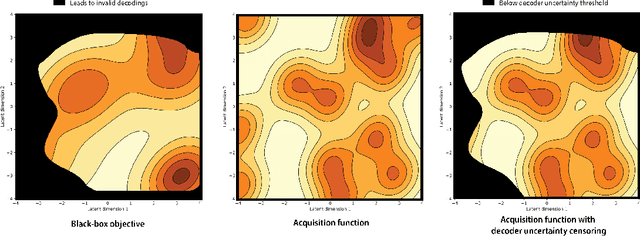
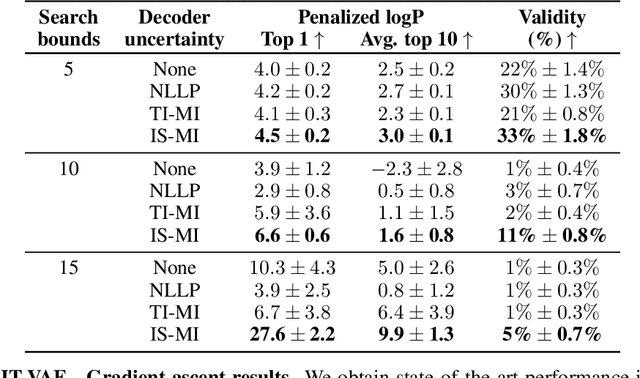


Optimization in the latent space of variational autoencoders is a promising approach to generate high-dimensional discrete objects that maximize an expensive black-box property (e.g., drug-likeness in molecular generation, function approximation with arithmetic expressions). However, existing methods lack robustness as they may decide to explore areas of the latent space for which no data was available during training and where the decoder can be unreliable, leading to the generation of unrealistic or invalid objects. We propose to leverage the epistemic uncertainty of the decoder to guide the optimization process. This is not trivial though, as a naive estimation of uncertainty in the high-dimensional and structured settings we consider would result in high estimator variance. To solve this problem, we introduce an importance sampling-based estimator that provides more robust estimates of epistemic uncertainty. Our uncertainty-guided optimization approach does not require modifications of the model architecture nor the training process. It produces samples with a better trade-off between black-box objective and validity of the generated samples, sometimes improving both simultaneously. We illustrate these advantages across several experimental settings in digit generation, arithmetic expression approximation and molecule generation for drug design.
SliceOut: Training Transformers and CNNs faster while using less memory
Jul 21, 2020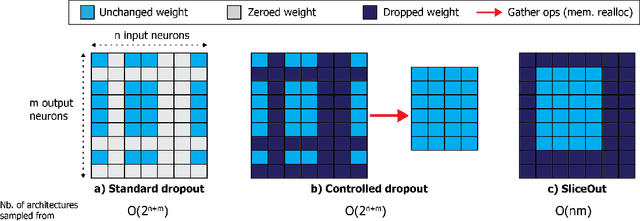
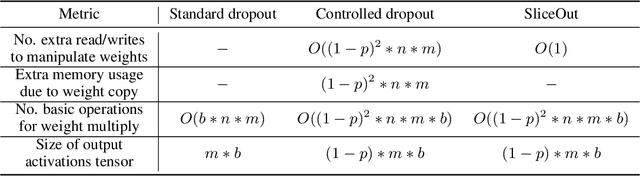
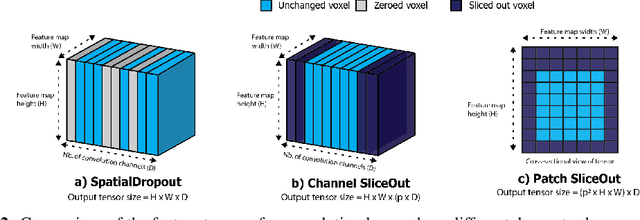
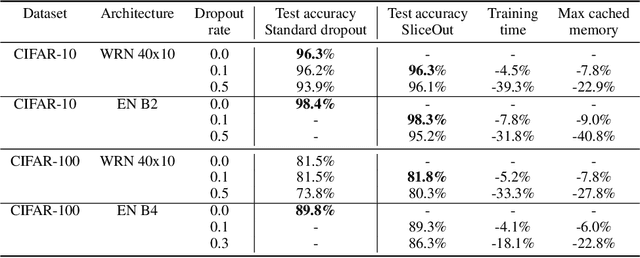
We demonstrate 10-40% speedups and memory reduction with Wide ResNets, EfficientNets, and Transformer models, with minimal to no loss in accuracy, using SliceOut---a new dropout scheme designed to take advantage of GPU memory layout. By dropping contiguous sets of units at random, our method preserves the regularization properties of dropout while allowing for more efficient low-level implementation, resulting in training speedups through (1) fast memory access and matrix multiplication of smaller tensors, and (2) memory savings by avoiding allocating memory to zero units in weight gradients and activations. Despite its simplicity, our method is highly effective. We demonstrate its efficacy at scale with Wide ResNets & EfficientNets on CIFAR10/100 and ImageNet, as well as Transformers on the LM1B dataset. These speedups and memory savings in training can lead to $CO_2$ emissions reduction of up to 40% for training large models.