 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTowards Diverse Scientific Hypothesis Search with Large Language Models
Jun 09, 2026Large language models (LLMs) are on the rise for accelerating scientific discovery, most recently in advanced tasks such as generating valid scientific hypotheses. Yet in many discovery settings, the goal is not to identify a single best hypothesis since validation can be noisy and expensive, and scientists benefit from a set of high-quality alternative hypotheses that hedge against downstream uncertainty for the best solutions. Nevertheless, commonly used evolutionary search recipes tend to prioritize optimization over exploration in hypothesis generation, and the resulting selection pressure during the search process leads to diversity collapse. Motivated by these limitations, we formulate hypothesis search as a sampling problem, where the objective is to efficiently produce diverse, high-quality hypotheses under a fixed validation budget. Building on this perspective, we propose \ours, an evolutionary framework inspired by the classical parallel tempering algorithm that searches hypotheses at multiple temperature levels and enables principled information exchange across temperatures to improve exploration without disrupting convergence. Across domains including molecular discovery, equation discovery, and algorithm discovery, our approach consistently improves both hypothesis quality and diversity under the same validation budget, and produces candidates that remain robust under more expensive downstream computational validations.
Free energy Estimation on Any State Space
May 29, 2026Free energy estimation is a fundamental yet challenging problem, from physics to statistics. Classical approaches rely on thermodynamic transformations, ranging from direct estimation, quasistatic integration, to finite-time averaging. Recent work [He and Du et al., 2025] learns neural transports to significantly accelerate the efficiency in the finite-time regime. In this paper, we generalize this framework to arbitrary state spaces. Building on this view, we develop a generalized neural transport learning approach for efficient estimation. Experiments validate the effectiveness and efficiency of the proposed method beyond continuous settings, extending to discrete and multimodal spaces as well as autoregressive settings. Beyond free energy estimation, we establish algebraic identities and reveal a group-theoretic structure linking infinitesimal time reversal and generalized Doob's $h$-transforms, showing that their compositions form a generalized dihedral group.
Decoupled PFNs: Identifiable Epistemic-Aleatoric Decomposition via Structured Synthetic Priors
May 07, 2026Prior-Fitted Networks (PFNs) amortize Bayesian prediction by meta-learning over a synthetic task prior, but their standard output is a posterior predictive distribution over noisy observations. For sequential decision-making, such as active learning and Bayesian optimization, acquisition should prioritize epistemic uncertainty about the latent signal rather than irreducible aleatoric observation noise. We show that this epistemic--aleatoric split is not identifiable in general from the posterior predictive distribution alone, even when that distribution is known exactly. We then exploit a distinctive advantage of PFNs: because the synthetic data-generating process is under our control, each task can contain an explicit latent signal and noise function, and the generator can provide query-level labels for both the noiseless target and the observation-noise variance. We use these labels to train a decoupled PFN with separate latent-signal and aleatoric heads. The observation-level predictive is induced by convolving the latent signal distribution with the learned noise model. Empirically, epistemic-only acquisition mitigates the failure mode of total-variance exploration in noisy and heteroscedastic settings. In matched comparisons, decoupled models usually improve over tuned observation-level baselines, with the clearest gains in HPO; in broader sweeps, a decoupled model obtains the best average rank in both HPO and synthetic BO.
Conditional Diffusion Sampling
May 05, 2026Sampling from unnormalized multimodal distributions with limited density evaluations remains a fundamental challenge in machine learning and natural sciences. Successful approaches construct a bridge between a tractable reference and the target distribution. Parallel Tempering (PT) serves as the gold standard, while recent diffusion-based approaches offer a continuous alternative at the cost of neural training. In this work, we introduce Conditional Diffusion Sampling (CDS), a framework that combines these two paradigms. To this end, we derive Conditional Interpolants, a class of stochastic processes whose transport dynamics are governed by an exact, closed-form stochastic differential equation (SDE), requiring no neural approximation. Although these dynamics require sampling from a non-trivial initialization distribution, we show both theoretically and empirically that the cost of this initialization diminishes for sufficiently short diffusion times. CDS leverages this by a two-stage procedure: (1) PT is used to efficiently sample the initial distribution, and then (2) samples are transported via the transport SDE. This combination couples the robust global exploration of PT with efficient local transport. Experiments suggest that CDS has the potential to achieve a superior trade-off between sample quality and density evaluation cost compared to state-of-the-art samplers.
Wiener Chaos Expansion based Neural Operator for Singular Stochastic Partial Differential Equations
Mar 09, 2026In this paper, we explore how our recently developed Wiener Chaos Expansion (WCE)-based neural operator (NO) can be applied to singular stochastic partial differential equations, e.g., the dynamic $\boldsymbolΦ^4_2$ model simulated in the recent works. Unlike the previous WCE-NO which solves SPDEs by simply inserting Wick-Hermite features into the backbone NO model, we leverage feature-wise linear modulation (FiLM) to appropriately capture the dependency between the solution of singular SPDE and its smooth remainder. The resulting WCE-FiLM-NO shows excellent performance on $\boldsymbolΦ^4_2$, as measured by relative $L_2$ loss, out-of-distribution $L_2$ loss, and autocorrelation score; all without the help of renormalisation factor. In addition, we also show the potential of simulating $\boldsymbolΦ^4_3$ data, which is more aligned with real scientific practice in statistical quantum field theory. To the best of our knowledge, this is among the first works to develop an efficient data-driven surrogate for the dynamical $\boldsymbolΦ^4_3$ model.
Compression as Adaptation: Implicit Visual Representation with Diffusion Foundation Models
Mar 08, 2026Modern visual generative models acquire rich visual knowledge through large-scale training, yet existing visual representations (such as pixels, latents, or tokens) remain external to the model and cannot directly exploit this knowledge for compact storage or reuse. In this work, we introduce a new visual representation framework that encodes a signal as a function, which is parametrized by low-rank adaptations attached to a frozen visual generative model. Such implicit representations of visual signals, \textit{e.g.}, an 81-frame video, can further be hashed into a single compact vector, achieving strong perceptual video compression at extremely low bitrates. Beyond basic compression, the functional nature of this representation enables inference-time scaling and control, allowing additional refinement on the compression performance. More broadly, as the implicit representations directly act as a function of the generation process, this suggests a unified framework bridging visual compression and generation.
Stochastic Interpolants in Hilbert Spaces
Feb 02, 2026Although diffusion models have successfully extended to function-valued data, stochastic interpolants -- which offer a flexible way to bridge arbitrary distributions -- remain limited to finite-dimensional settings. This work bridges this gap by establishing a rigorous framework for stochastic interpolants in infinite-dimensional Hilbert spaces. We provide comprehensive theoretical foundations, including proofs of well-posedness and explicit error bounds. We demonstrate the effectiveness of the proposed framework for conditional generation, focusing particularly on complex PDE-based benchmarks. By enabling generative bridges between arbitrary functional distributions, our approach achieves state-of-the-art results, offering a powerful, general-purpose tool for scientific discovery.
A Diffusive Classification Loss for Learning Energy-based Generative Models
Jan 28, 2026Score-based generative models have recently achieved remarkable success. While they are usually parameterized by the score, an alternative way is to use a series of time-dependent energy-based models (EBMs), where the score is obtained from the negative input-gradient of the energy. Crucially, EBMs can be leveraged not only for generation, but also for tasks such as compositional sampling or building Boltzmann Generators via Monte Carlo methods. However, training EBMs remains challenging. Direct maximum likelihood is computationally prohibitive due to the need for nested sampling, while score matching, though efficient, suffers from mode blindness. To address these issues, we introduce the Diffusive Classification (DiffCLF) objective, a simple method that avoids blindness while remaining computationally efficient. DiffCLF reframes EBM learning as a supervised classification problem across noise levels, and can be seamlessly combined with standard score-based objectives. We validate the effectiveness of DiffCLF by comparing the estimated energies against ground truth in analytical Gaussian mixture cases, and by applying the trained models to tasks such as model composition and Boltzmann Generator sampling. Our results show that DiffCLF enables EBMs with higher fidelity and broader applicability than existing approaches.
Expanding the Chaos: Neural Operator for Stochastic (Partial) Differential Equations
Jan 03, 2026Stochastic differential equations (SDEs) and stochastic partial differential equations (SPDEs) are fundamental tools for modeling stochastic dynamics across the natural sciences and modern machine learning. Developing deep learning models for approximating their solution operators promises not only fast, practical solvers, but may also inspire models that resolve classical learning tasks from a new perspective. In this work, we build on classical Wiener chaos expansions (WCE) to design neural operator (NO) architectures for SPDEs and SDEs: we project the driving noise paths onto orthonormal Wick Hermite features and parameterize the resulting deterministic chaos coefficients with neural operators, so that full solution trajectories can be reconstructed from noise in a single forward pass. On the theoretical side, we investigate the classical WCE results for the class of multi-dimensional SDEs and semilinear SPDEs considered here by explicitly writing down the associated coupled ODE/PDE systems for their chaos coefficients, which makes the separation between stochastic forcing and deterministic dynamics fully explicit and directly motivates our model designs. On the empirical side, we validate our models on a diverse suite of problems: classical SPDE benchmarks, diffusion one-step sampling on images, topological interpolation on graphs, financial extrapolation, parameter estimation, and manifold SDEs for flood prediction, demonstrating competitive accuracy and broad applicability. Overall, our results indicate that WCE-based neural operators provide a practical and scalable way to learn SDE/SPDE solution operators across diverse domains.
Scalable Gaussian Processes with Latent Kronecker Structure
Jun 07, 2025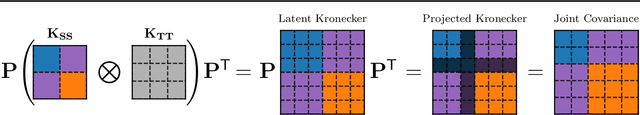
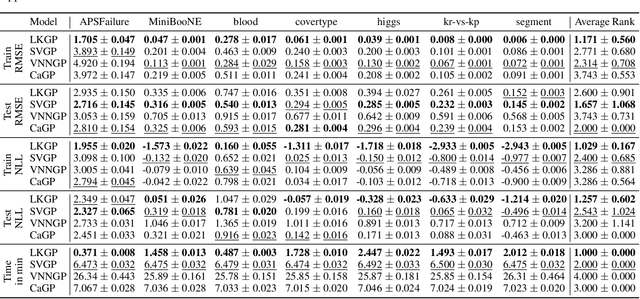
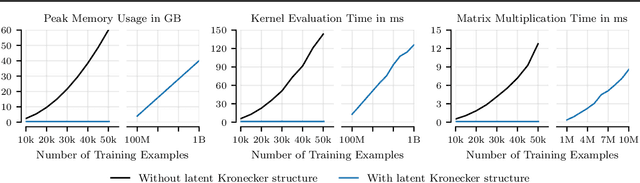
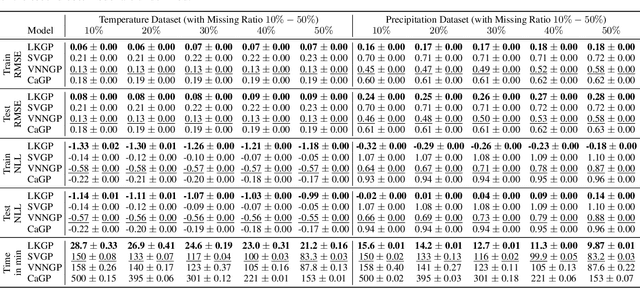
Applying Gaussian processes (GPs) to very large datasets remains a challenge due to limited computational scalability. Matrix structures, such as the Kronecker product, can accelerate operations significantly, but their application commonly entails approximations or unrealistic assumptions. In particular, the most common path to creating a Kronecker-structured kernel matrix is by evaluating a product kernel on gridded inputs that can be expressed as a Cartesian product. However, this structure is lost if any observation is missing, breaking the Cartesian product structure, which frequently occurs in real-world data such as time series. To address this limitation, we propose leveraging latent Kronecker structure, by expressing the kernel matrix of observed values as the projection of a latent Kronecker product. In combination with iterative linear system solvers and pathwise conditioning, our method facilitates inference of exact GPs while requiring substantially fewer computational resources than standard iterative methods. We demonstrate that our method outperforms state-of-the-art sparse and variational GPs on real-world datasets with up to five million examples, including robotics, automated machine learning, and climate applications.