 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIn-silico biological discovery with large perturbation models
Mar 30, 2025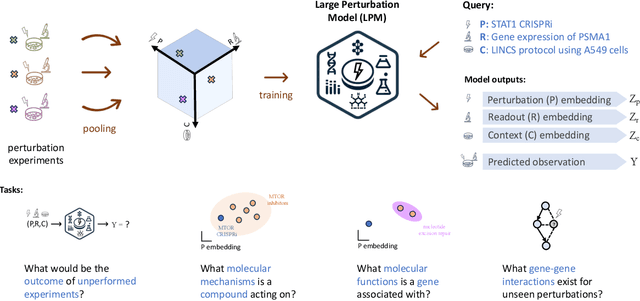
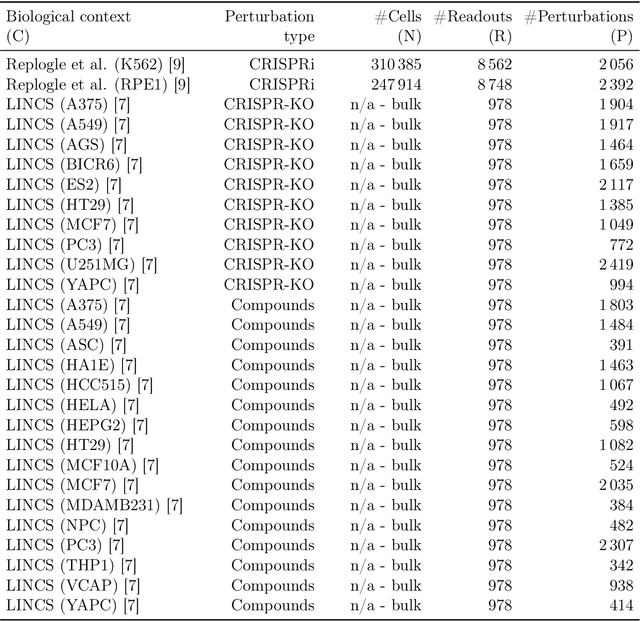
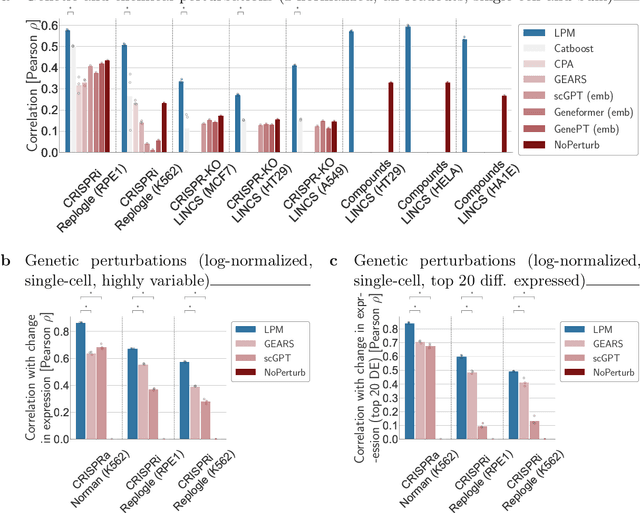
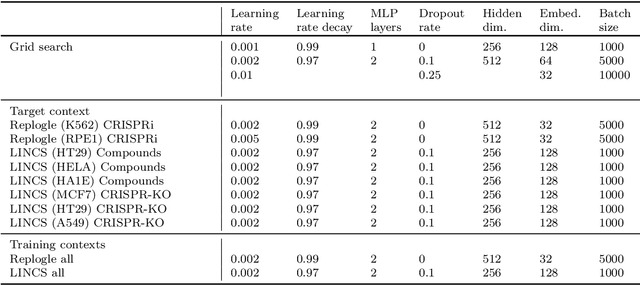
Data generated in perturbation experiments link perturbations to the changes they elicit and therefore contain information relevant to numerous biological discovery tasks -- from understanding the relationships between biological entities to developing therapeutics. However, these data encompass diverse perturbations and readouts, and the complex dependence of experimental outcomes on their biological context makes it challenging to integrate insights across experiments. Here, we present the Large Perturbation Model (LPM), a deep-learning model that integrates multiple, heterogeneous perturbation experiments by representing perturbation, readout, and context as disentangled dimensions. LPM outperforms existing methods across multiple biological discovery tasks, including in predicting post-perturbation transcriptomes of unseen experiments, identifying shared molecular mechanisms of action between chemical and genetic perturbations, and facilitating the inference of gene-gene interaction networks.
Multi-omics Prediction from High-content Cellular Imaging with Deep Learning
Jun 19, 2023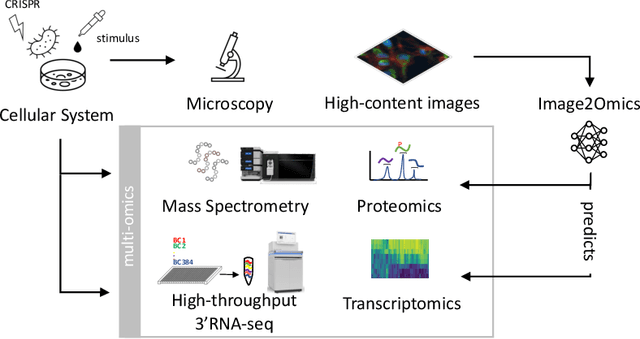
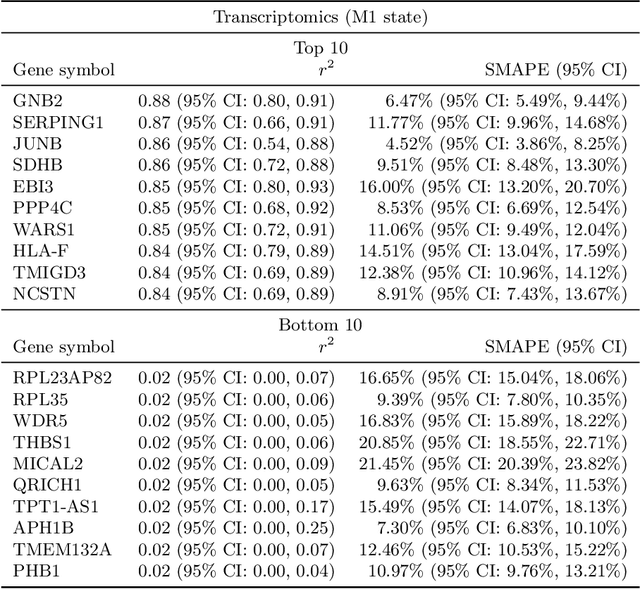
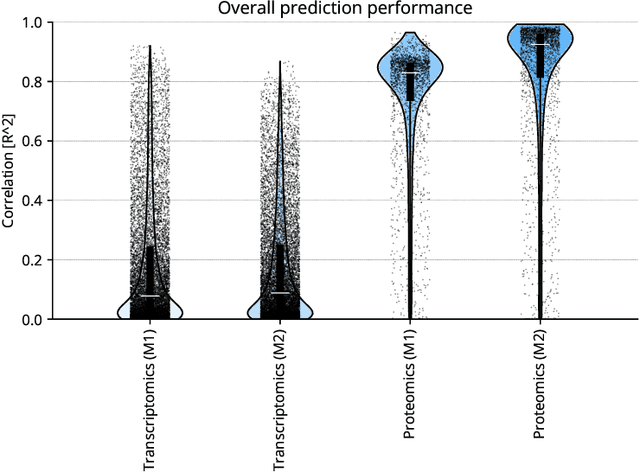
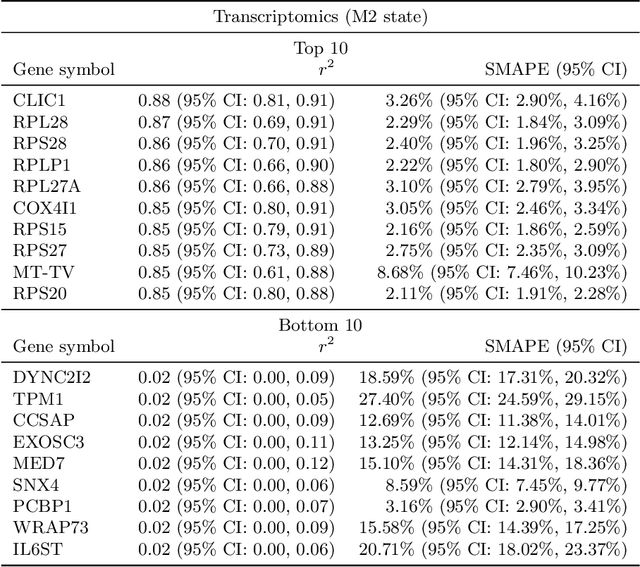
High-content cellular imaging, transcriptomics, and proteomics data provide rich and complementary views on the molecular layers of biology that influence cellular states and function. However, the biological determinants through which changes in multi-omics measurements influence cellular morphology have not yet been systematically explored, and the degree to which cell imaging could potentially enable the prediction of multi-omics directly from cell imaging data is therefore currently unclear. Here, we address the question of whether it is possible to predict bulk multi-omics measurements directly from cell images using Image2Omics -- a deep learning approach that predicts multi-omics in a cell population directly from high-content images stained with multiplexed fluorescent dyes. We perform an experimental evaluation in gene-edited macrophages derived from human induced pluripotent stem cell (hiPSC) under multiple stimulation conditions and demonstrate that Image2Omics achieves significantly better performance in predicting transcriptomics and proteomics measurements directly from cell images than predictors based on the mean observed training set abundance. We observed significant predictability of abundances for 5903 (22.43%; 95% CI: 8.77%, 38.88%) and 5819 (22.11%; 95% CI: 10.40%, 38.08%) transcripts out of 26137 in M1 and M2-stimulated macrophages respectively and for 1933 (38.77%; 95% CI: 36.94%, 39.85%) and 2055 (41.22%; 95% CI: 39.31%, 42.42%) proteins out of 4986 in M1 and M2-stimulated macrophages respectively. Our results show that some transcript and protein abundances are predictable from cell imaging and that cell imaging may potentially, in some settings and depending on the mechanisms of interest and desired performance threshold, even be a scalable and resource-efficient substitute for multi-omics measurements.