 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTemporal Convolutional Autoencoder for Interference Mitigation in FMCW Radar Altimeters
May 28, 2025

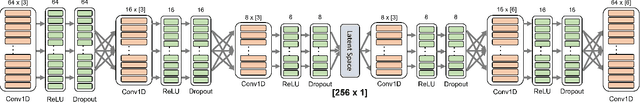

We investigate the end-to-end altitude estimation performance of a convolutional autoencoder-based interference mitigation approach for frequency-modulated continuous-wave (FMCW) radar altimeters. Specifically, we show that a Temporal Convolutional Network (TCN) autoencoder effectively exploits temporal correlations in the received signal, providing superior interference suppression compared to a Least Mean Squares (LMS) adaptive filter. Unlike existing approaches, the present method operates directly on the received FMCW signal. Additionally, we identify key challenges in applying deep learning to wideband FMCW interference mitigation and outline directions for future research to enhance real-time feasibility and generalization to arbitrary interference conditions.
Aircraft Radar Altimeter Interference Mitigation Through a CNN-Layer Only Denoising Autoencoder Architecture
Oct 04, 2024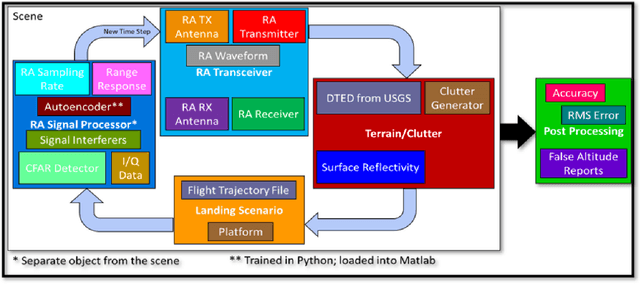
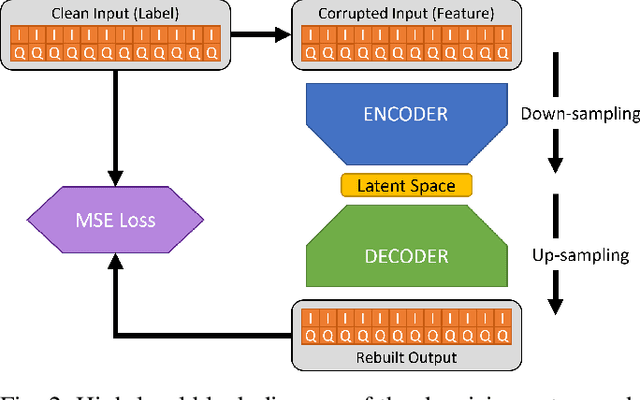
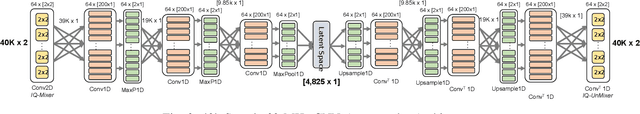
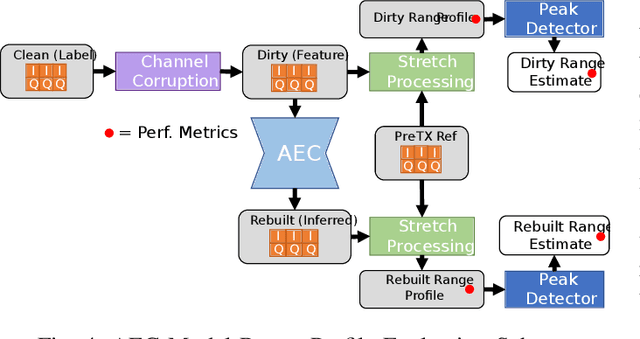
Denoising autoencoders for signal processing applications have been shown to experience significant difficulty in learning to reconstruct radio frequency communication signals, particularly in the large sample regime. In communication systems, this challenge is primarily due to the need to reconstruct the modulated data stream which is generally highly stochastic in nature. In this work, we take advantage of this limitation by using the denoising autoencoder to instead remove interfering radio frequency communication signals while reconstructing highly structured FMCW radar signals. More specifically, in this work we show that a CNN-layer only autoencoder architecture can be utilized to improve the accuracy of a radar altimeter's ranging estimate even in severe interference environments consisting of a multitude of interference signals. This is demonstrated through comprehensive performance analysis of an end-to-end FMCW radar altimeter simulation with and without the convolutional layer-only autoencoder. The proposed approach significantly improves interference mitigation in the presence of both narrow-band tone interference as well as wideband QPSK interference in terms of range RMS error, number of false altitude reports, and the peak-to-sidelobe ratio of the resulting range profile. FMCW radar signals of up to 40,000 IQ samples can be reliably reconstructed.
Fine-tuning Protein Language Models with Deep Mutational Scanning improves Variant Effect Prediction
May 10, 2024
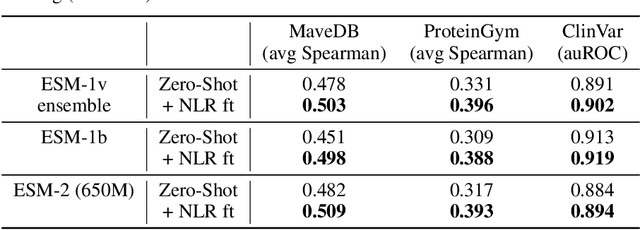

Protein Language Models (PLMs) have emerged as performant and scalable tools for predicting the functional impact and clinical significance of protein-coding variants, but they still lag experimental accuracy. Here, we present a novel fine-tuning approach to improve the performance of PLMs with experimental maps of variant effects from Deep Mutational Scanning (DMS) assays using a Normalised Log-odds Ratio (NLR) head. We find consistent improvements in a held-out protein test set, and on independent DMS and clinical variant annotation benchmarks from ProteinGym and ClinVar. These findings demonstrate that DMS is a promising source of sequence diversity and supervised training data for improving the performance of PLMs for variant effect prediction.
Generalising sequence models for epigenome predictions with tissue and assay embeddings
Aug 22, 2023Sequence modelling approaches for epigenetic profile prediction have recently expanded in terms of sequence length, model size, and profile diversity. However, current models cannot infer on many experimentally feasible tissue and assay pairs due to poor usage of contextual information, limiting $\textit{in silico}$ understanding of regulatory genomics. We demonstrate that strong correlation can be achieved across a large range of experimental conditions by integrating tissue and assay embeddings into a Contextualised Genomic Network (CGN). In contrast to previous approaches, we enhance long-range sequence embeddings with contextual information in the input space, rather than expanding the output space. We exhibit the efficacy of our approach across a broad set of epigenetic profiles and provide the first insights into the effect of genetic variants on epigenetic sequence model training. Our general approach to context integration exceeds state of the art in multiple settings while employing a more rigorous validation procedure.
Epigenomic language models powered by Cerebras
Dec 14, 2021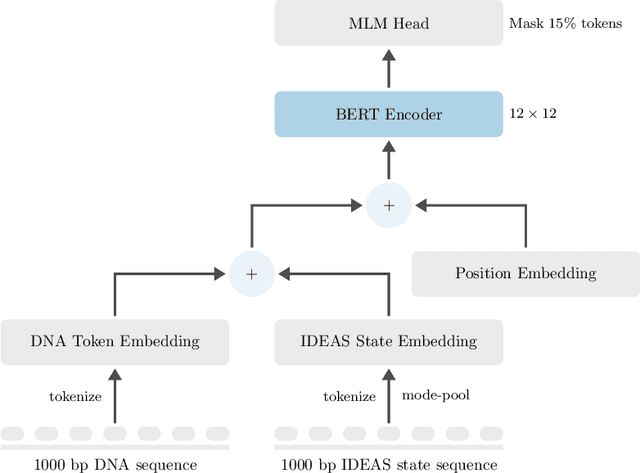
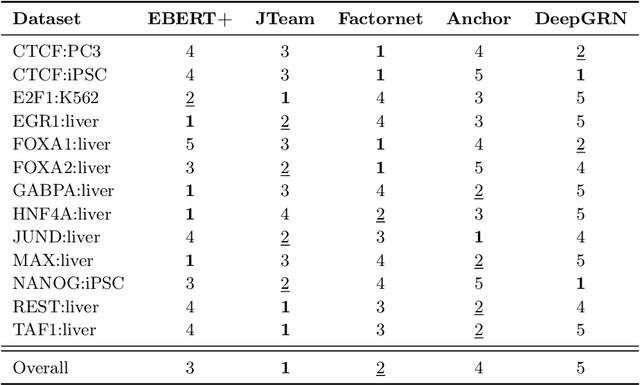
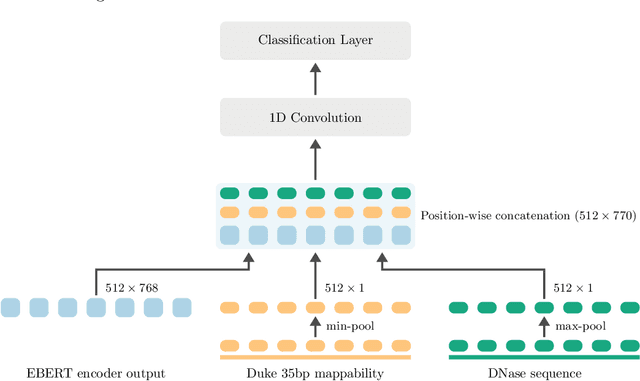
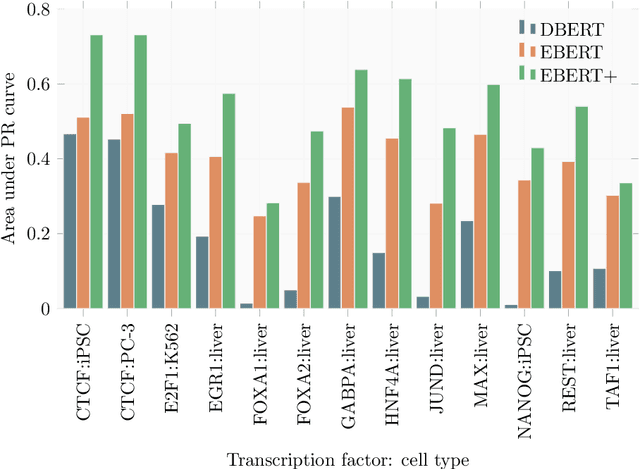
Large scale self-supervised pre-training of Transformer language models has advanced the field of Natural Language Processing and shown promise in cross-application to the biological `languages' of proteins and DNA. Learning effective representations of DNA sequences using large genomic sequence corpuses may accelerate the development of models of gene regulation and function through transfer learning. However, to accurately model cell type-specific gene regulation and function, it is necessary to consider not only the information contained in DNA nucleotide sequences, which is mostly invariant between cell types, but also how the local chemical and structural `epigenetic state' of chromosomes varies between cell types. Here, we introduce a Bidirectional Encoder Representations from Transformers (BERT) model that learns representations based on both DNA sequence and paired epigenetic state inputs, which we call Epigenomic BERT (or EBERT). We pre-train EBERT with a masked language model objective across the entire human genome and across 127 cell types. Training this complex model with a previously prohibitively large dataset was made possible for the first time by a partnership with Cerebras Systems, whose CS-1 system powered all pre-training experiments. We show EBERT's transfer learning potential by demonstrating strong performance on a cell type-specific transcription factor binding prediction task. Our fine-tuned model exceeds state of the art performance on 4 of 13 evaluation datasets from ENCODE-DREAM benchmarks and earns an overall rank of 3rd on the challenge leaderboard. We explore how the inclusion of epigenetic data and task specific feature augmentation impact transfer learning performance.