 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOut-of-distribution evaluations of channel agnostic masked autoencoders in fluorescence microscopy
Mar 24, 2025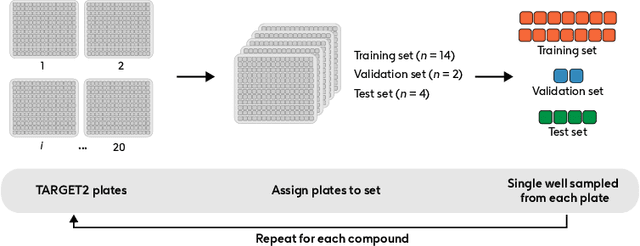
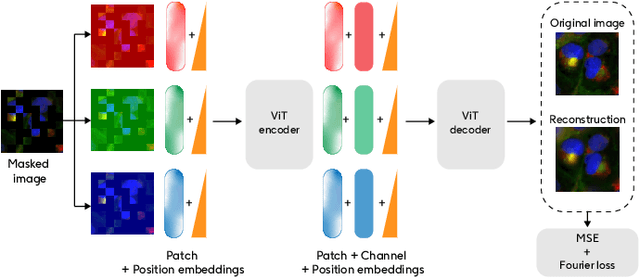
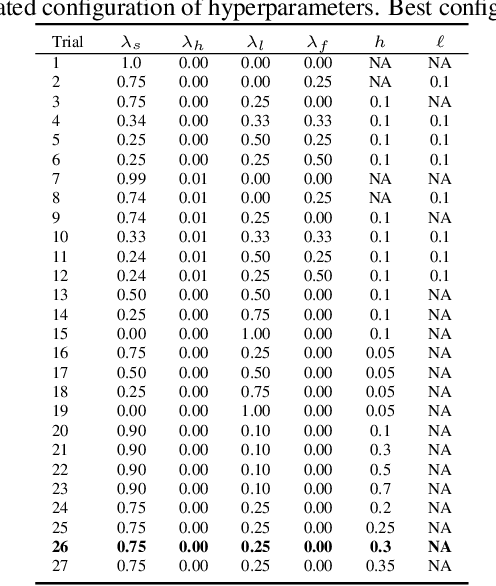
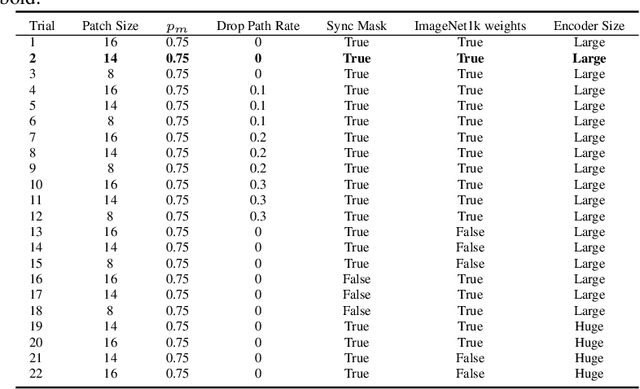
Developing computer vision for high-content screening is challenging due to various sources of distribution-shift caused by changes in experimental conditions, perturbagens, and fluorescent markers. The impact of different sources of distribution-shift are confounded in typical evaluations of models based on transfer learning, which limits interpretations of how changes to model design and training affect generalisation. We propose an evaluation scheme that isolates sources of distribution-shift using the JUMP-CP dataset, allowing researchers to evaluate generalisation with respect to specific sources of distribution-shift. We then present a channel-agnostic masked autoencoder $\mathbf{Campfire}$ which, via a shared decoder for all channels, scales effectively to datasets containing many different fluorescent markers, and show that it generalises to out-of-distribution experimental batches, perturbagens, and fluorescent markers, and also demonstrates successful transfer learning from one cell type to another.
Leak Proof CMap; a framework for training and evaluation of cell line agnostic L1000 similarity methods
Apr 29, 2024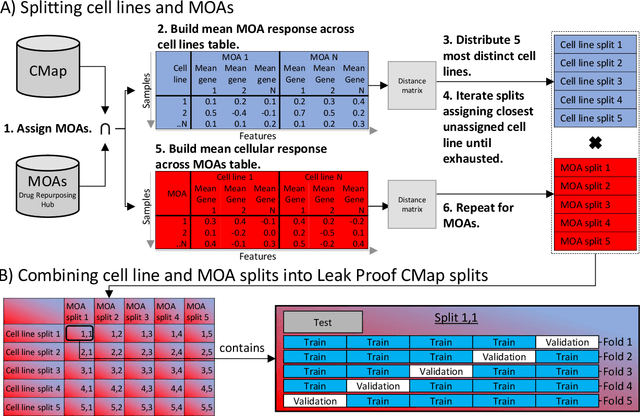
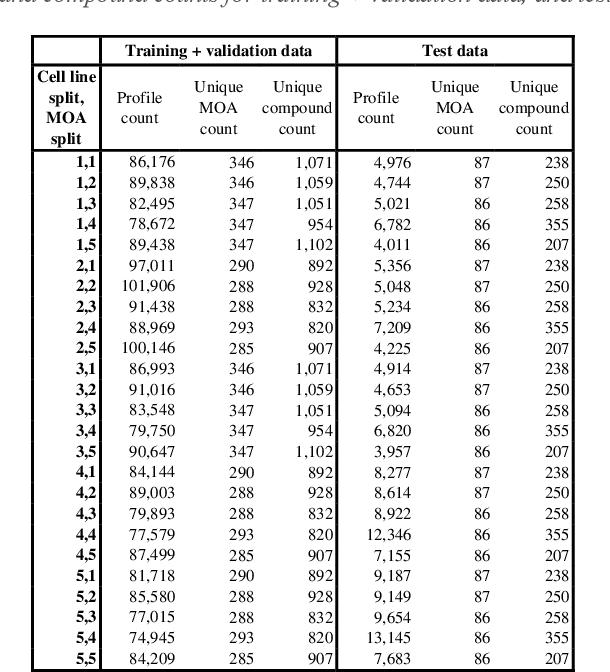
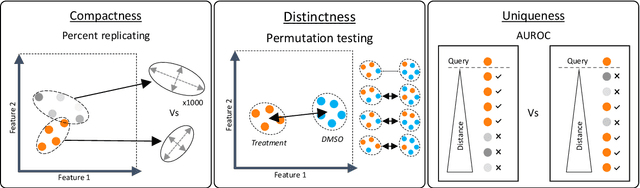
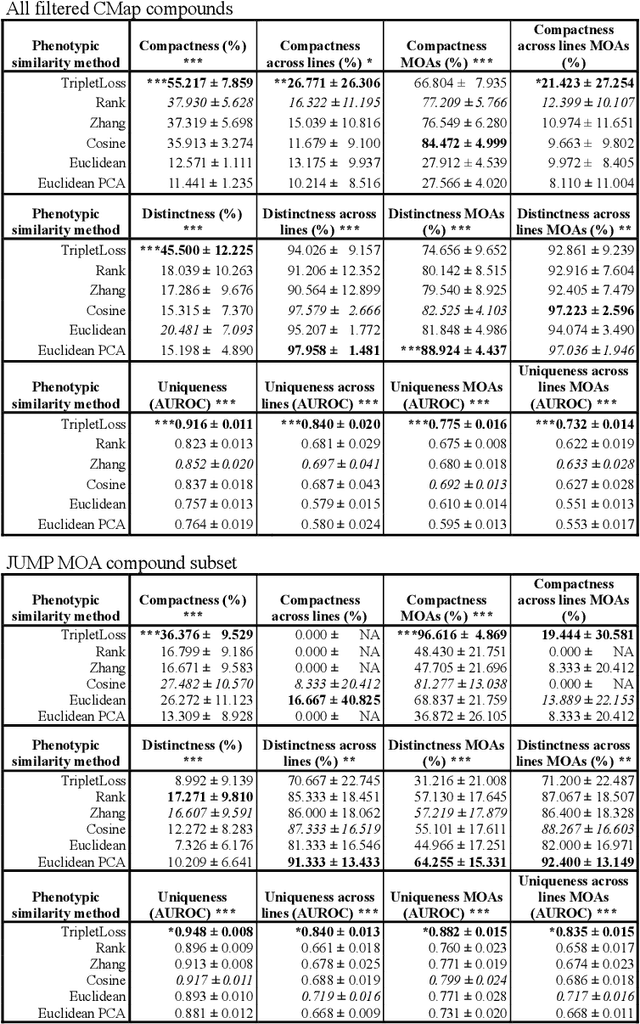
The Connectivity Map (CMap) is a large publicly available database of cellular transcriptomic responses to chemical and genetic perturbations built using a standardized acquisition protocol known as the L1000 technique. Databases such as CMap provide an exciting opportunity to enrich drug discovery efforts, providing a 'known' phenotypic landscape to explore and enabling the development of state of the art techniques for enhanced information extraction and better informed decisions. Whilst multiple methods for measuring phenotypic similarity and interrogating profiles have been developed, the field is severely lacking standardized benchmarks using appropriate data splitting for training and unbiased evaluation of machine learning methods. To address this, we have developed 'Leak Proof CMap' and exemplified its application to a set of common transcriptomic and generic phenotypic similarity methods along with an exemplar triplet loss-based method. Benchmarking in three critical performance areas (compactness, distinctness, and uniqueness) is conducted using carefully crafted data splits ensuring no similar cell lines or treatments with shared or closely matching responses or mechanisms of action are present in training, validation, or test sets. This enables testing of models with unseen samples akin to exploring treatments with novel modes of action in novel patient derived cell lines. With a carefully crafted benchmark and data splitting regime in place, the tooling now exists to create performant phenotypic similarity methods for use in personalized medicine (novel cell lines) and to better augment high throughput phenotypic screening technologies with the L1000 transcriptomic technology.
Enhancing Few-shot Image Classification with Cosine Transformer
Nov 16, 2022

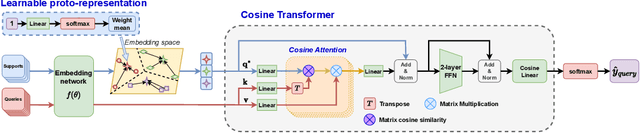
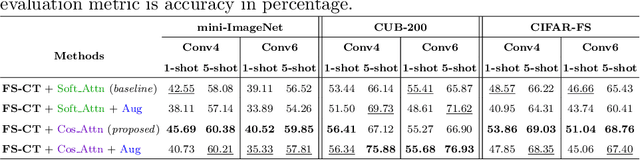
This paper addresses the few-shot image classification problem. One notable limitation of few-shot learning is the variation in describing the same category, which might result in a significant difference between small labeled support and large unlabeled query sets. Our approach is to obtain a relation heatmap between the two sets in order to label the latter one in a transductive setting manner. This can be solved by using cross-attention with the scaled dot-product mechanism. However, the magnitude differences between two separate sets of embedding vectors may cause a significant impact on the output attention map and affect model performance. We tackle this problem by improving the attention mechanism with cosine similarity. Specifically, we develop FS-CT (Few-shot Cosine Transformer), a few-shot image classification method based on prototypical embedding and transformer-based framework. The proposed Cosine attention improves FS-CT performances significantly from nearly 5% to over 20% in accuracy compared to the baseline scaled dot-product attention in various scenarios on three few-shot datasets mini-ImageNet, CUB-200, and CIFAR-FS. Additionally, we enhance the prototypical embedding for categorical representation with learnable weights before feeding them to the attention module. Our proposed method FS-CT along with the Cosine attention is simple to implement and can be applied for a wide range of applications. Our codes are available at https://github.com/vinuni-vishc/Few-Shot-Cosine-Transformer
Epigenomic language models powered by Cerebras
Dec 14, 2021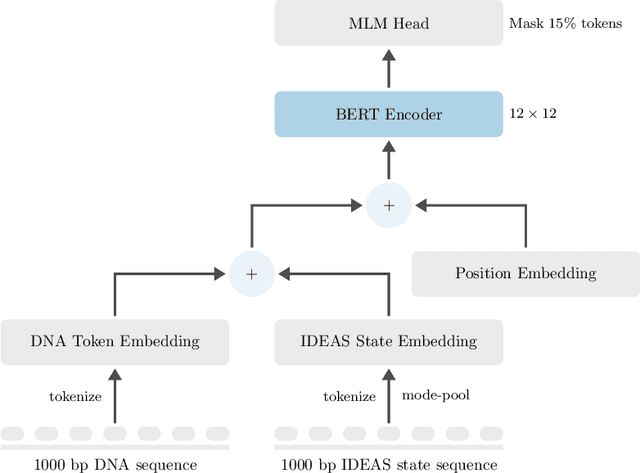
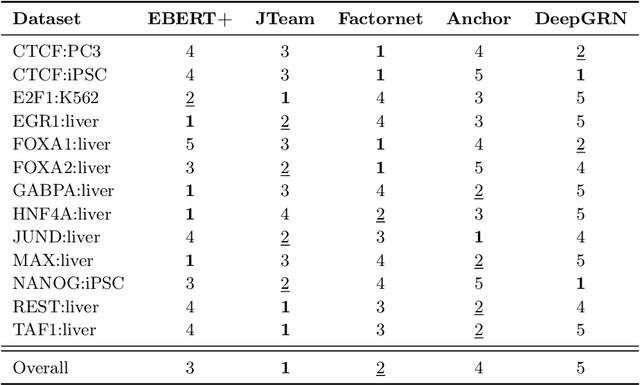
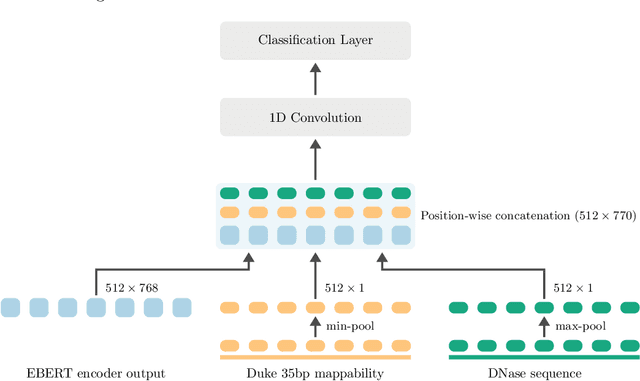
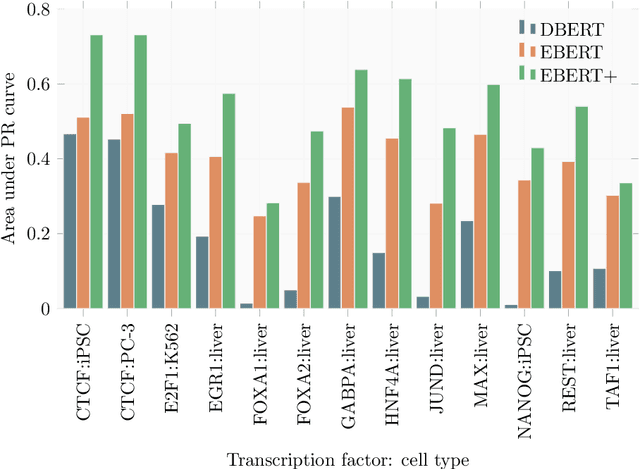
Large scale self-supervised pre-training of Transformer language models has advanced the field of Natural Language Processing and shown promise in cross-application to the biological `languages' of proteins and DNA. Learning effective representations of DNA sequences using large genomic sequence corpuses may accelerate the development of models of gene regulation and function through transfer learning. However, to accurately model cell type-specific gene regulation and function, it is necessary to consider not only the information contained in DNA nucleotide sequences, which is mostly invariant between cell types, but also how the local chemical and structural `epigenetic state' of chromosomes varies between cell types. Here, we introduce a Bidirectional Encoder Representations from Transformers (BERT) model that learns representations based on both DNA sequence and paired epigenetic state inputs, which we call Epigenomic BERT (or EBERT). We pre-train EBERT with a masked language model objective across the entire human genome and across 127 cell types. Training this complex model with a previously prohibitively large dataset was made possible for the first time by a partnership with Cerebras Systems, whose CS-1 system powered all pre-training experiments. We show EBERT's transfer learning potential by demonstrating strong performance on a cell type-specific transcription factor binding prediction task. Our fine-tuned model exceeds state of the art performance on 4 of 13 evaluation datasets from ENCODE-DREAM benchmarks and earns an overall rank of 3rd on the challenge leaderboard. We explore how the inclusion of epigenetic data and task specific feature augmentation impact transfer learning performance.
Meta-Learning Initializations for Low-Resource Drug Discovery
Mar 12, 2020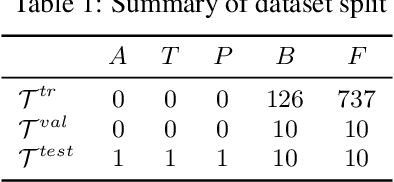
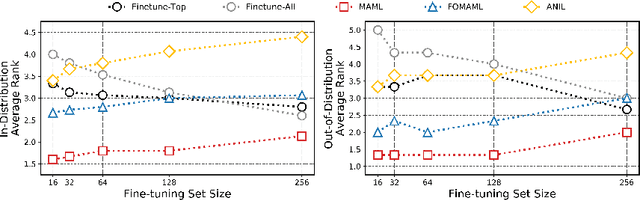
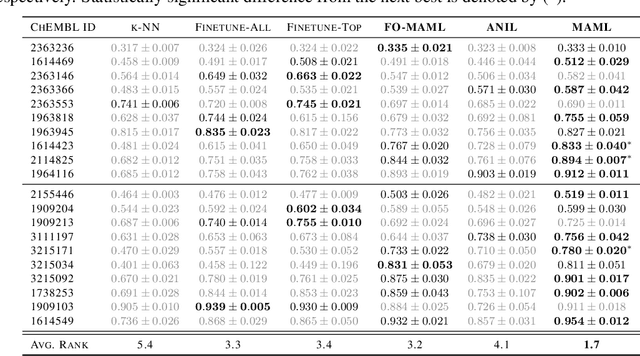
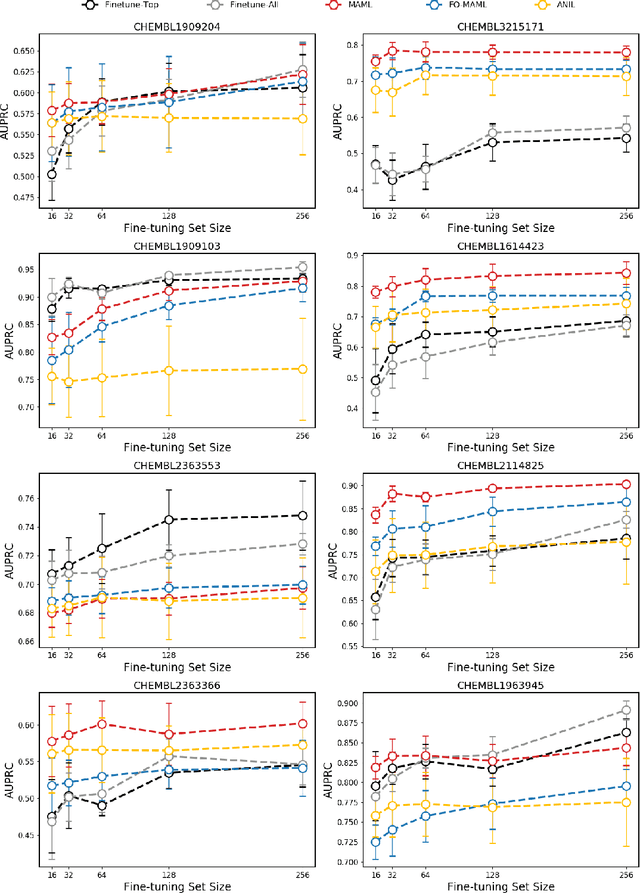
Building in silico models to predict chemical properties and activities is a crucial step in drug discovery. However, drug discovery projects are often characterized by limited labeled data, hindering the applications of deep learning in this setting. Meanwhile advances in meta-learning have enabled state-of-the-art performances in few-shot learning benchmarks, naturally prompting the question: Can meta-learning improve deep learning performance in low-resource drug discovery projects? In this work, we assess the efficiency of the Model-Agnostic Meta-Learning (MAML) algorithm - along with its variants FO-MAML and ANIL - at learning to predict chemical properties and activities. Using the ChEMBL20 dataset to emulate low-resource settings, our benchmark shows that meta-initializations perform comparably to or outperform multi-task pre-training baselines on 16 out of 20 in-distribution tasks and on all out-of-distribution tasks, providing an average improvement in AUPRC of 7.2% and 14.9% respectively. Finally, we observe that meta-initializations consistently result in the best performing models across fine-tuning sets with $k \in \{16, 32, 64, 128, 256\}$ instances.