 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFine-tuning Protein Language Models with Deep Mutational Scanning improves Variant Effect Prediction
May 10, 2024
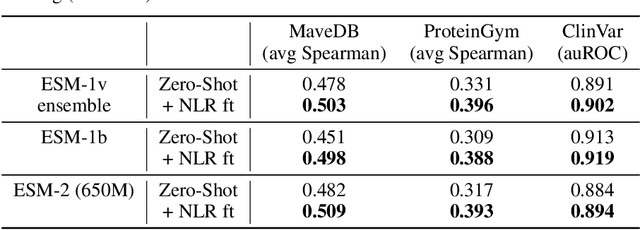

Protein Language Models (PLMs) have emerged as performant and scalable tools for predicting the functional impact and clinical significance of protein-coding variants, but they still lag experimental accuracy. Here, we present a novel fine-tuning approach to improve the performance of PLMs with experimental maps of variant effects from Deep Mutational Scanning (DMS) assays using a Normalised Log-odds Ratio (NLR) head. We find consistent improvements in a held-out protein test set, and on independent DMS and clinical variant annotation benchmarks from ProteinGym and ClinVar. These findings demonstrate that DMS is a promising source of sequence diversity and supervised training data for improving the performance of PLMs for variant effect prediction.
Generalising sequence models for epigenome predictions with tissue and assay embeddings
Aug 22, 2023Sequence modelling approaches for epigenetic profile prediction have recently expanded in terms of sequence length, model size, and profile diversity. However, current models cannot infer on many experimentally feasible tissue and assay pairs due to poor usage of contextual information, limiting $\textit{in silico}$ understanding of regulatory genomics. We demonstrate that strong correlation can be achieved across a large range of experimental conditions by integrating tissue and assay embeddings into a Contextualised Genomic Network (CGN). In contrast to previous approaches, we enhance long-range sequence embeddings with contextual information in the input space, rather than expanding the output space. We exhibit the efficacy of our approach across a broad set of epigenetic profiles and provide the first insights into the effect of genetic variants on epigenetic sequence model training. Our general approach to context integration exceeds state of the art in multiple settings while employing a more rigorous validation procedure.