 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnzyPGM: Pocket-conditioned Generative Model for Substrate-specific Enzyme Design
Jan 27, 2026Designing enzymes with substrate-binding pockets is a critical challenge in protein engineering, as catalytic activity depends on the precise interaction between pockets and substrates. Currently, generative models dominate functional protein design but cannot model pocket-substrate interactions, which limits the generation of enzymes with precise catalytic environments. To address this issue, we propose EnzyPGM, a unified framework that jointly generates enzymes and substrate-binding pockets conditioned on functional priors and substrates, with a particular focus on learning accurate pocket-substrate interactions. At its core, EnzyPGM includes two main modules: a Residue-atom Bi-scale Attention (RBA) that jointly models intra-residue dependencies and fine-grained interactions between pocket residues and substrate atoms, and a Residue Function Fusion (RFF) that incorporates enzyme function priors into residue representations. Also, we curate EnzyPock, an enzyme-pocket dataset comprising 83,062 enzyme-substrate pairs across 1,036 four-level enzyme families. Extensive experiments demonstrate that EnzyPGM achieves state-of-the-art performance on EnzyPock. Notably, EnzyPGM reduces the average binding energy of 0.47 kcal/mol over EnzyGen, showing its superior performance on substrate-specific enzyme design. The code and dataset will be released later.
Beyond Affinity: A Benchmark of 1D, 2D, and 3D Methods Reveals Critical Trade-offs in Structure-Based Drug Design
Jan 13, 2026Currently, the field of structure-based drug design is dominated by three main types of algorithms: search-based algorithms, deep generative models, and reinforcement learning. While existing works have typically focused on comparing models within a single algorithmic category, cross-algorithm comparisons remain scarce. In this paper, to fill the gap, we establish a benchmark to evaluate the performance of fifteen models across these different algorithmic foundations by assessing the pharmaceutical properties of the generated molecules and their docking affinities and poses with specified target proteins. We highlight the unique advantages of each algorithmic approach and offer recommendations for the design of future SBDD models. We emphasize that 1D/2D ligand-centric drug design methods can be used in SBDD by treating the docking function as a black-box oracle, which is typically neglected. Our evaluation reveals distinct patterns across model categories. 3D structure-based models excel in binding affinities but show inconsistencies in chemical validity and pose quality. 1D models demonstrate reliable performance in standard molecular metrics but rarely achieve optimal binding affinities. 2D models offer balanced performance, maintaining high chemical validity while achieving moderate binding scores. Through detailed analysis across multiple protein targets, we identify key improvement areas for each model category, providing insights for researchers to combine strengths of different approaches while addressing their limitations. All the code that are used for benchmarking is available in https://github.com/zkysfls/2025-sbdd-benchmark
Quantized SO(3)-Equivariant Graph Neural Networks for Efficient Molecular Property Prediction
Jan 05, 2026Deploying 3D graph neural networks (GNNs) that are equivariant to 3D rotations (the group SO(3)) on edge devices is challenging due to their high computational cost. This paper addresses the problem by compressing and accelerating an SO(3)-equivariant GNN using low-bit quantization techniques. Specifically, we introduce three innovations for quantized equivariant transformers: (1) a magnitude-direction decoupled quantization scheme that separately quantizes the norm and orientation of equivariant (vector) features, (2) a branch-separated quantization-aware training strategy that treats invariant and equivariant feature channels differently in an attention-based $SO(3)$-GNN, and (3) a robustness-enhancing attention normalization mechanism that stabilizes low-precision attention computations. Experiments on the QM9 and rMD17 molecular benchmarks demonstrate that our 8-bit models achieve accuracy on energy and force predictions comparable to full-precision baselines with markedly improved efficiency. We also conduct ablation studies to quantify the contribution of each component to maintain accuracy and equivariance under quantization, using the Local error of equivariance (LEE) metric. The proposed techniques enable the deployment of symmetry-aware GNNs in practical chemistry applications with 2.37--2.73x faster inference and 4x smaller model size, without sacrificing accuracy or physical symmetry.
ClinicalReTrial: A Self-Evolving AI Agent for Clinical Trial Protocol Optimization
Jan 01, 2026Clinical trial failure remains a central bottleneck in drug development, where minor protocol design flaws can irreversibly compromise outcomes despite promising therapeutics. Although cutting-edge AI methods achieve strong performance in predicting trial success, they are inherently reactive for merely diagnosing risk without offering actionable remedies once failure is anticipated. To fill this gap, this paper proposes ClinicalReTrial, a self-evolving AI agent framework that addresses this gap by casting clinical trial reasoning as an iterative protocol redesign problem. Our method integrates failure diagnosis, safety-aware modification, and candidate evaluation in a closed-loop, reward-driven optimization framework. Serving the outcome prediction model as a simulation environment, ClinicalReTrial enables low-cost evaluation of protocol modifications and provides dense reward signals for continuous self-improvement. To support efficient exploration, the framework maintains hierarchical memory that captures iteration-level feedback within trials and distills transferable redesign patterns across trials. Empirically, ClinicalReTrial improves 83.3% of trial protocols with a mean success probability gain of 5.7%, and retrospective case studies demonstrate strong alignment between the discovered redesign strategies and real-world clinical trial modifications.
MolAct: An Agentic RL Framework for Molecular Editing and Property Optimization
Dec 24, 2025Molecular editing and optimization are multi-step problems that require iteratively improving properties while keeping molecules chemically valid and structurally similar. We frame both tasks as sequential, tool-guided decisions and introduce MolAct, an agentic reinforcement learning framework that employs a two-stage training paradigm: first building editing capability, then optimizing properties while reusing the learned editing behaviors. To the best of our knowledge, this is the first work to formalize molecular design as an Agentic Reinforcement Learning problem, where an LLM agent learns to interleave reasoning, tool-use, and molecular optimization. The framework enables agents to interact in multiple turns, invoking chemical tools for validity checking, property assessment, and similarity control, and leverages their feedback to refine subsequent edits. We instantiate the MolAct framework to train two model families: MolEditAgent for molecular editing tasks and MolOptAgent for molecular optimization tasks. In molecular editing, MolEditAgent-7B delivers 100, 95, and 98 valid add, delete, and substitute edits, outperforming strong closed "thinking" baselines such as DeepSeek-R1; MolEditAgent-3B approaches the performance of much larger open "thinking" models like Qwen3-32B-think. In molecular optimization, MolOptAgent-7B (trained on MolEditAgent-7B) surpasses the best closed "thinking" baseline (e.g., Claude 3.7) on LogP and remains competitive on solubility, while maintaining balanced performance across other objectives. These results highlight that treating molecular design as a multi-step, tool-augmented process is key to reliable and interpretable improvements.
Probing Scientific General Intelligence of LLMs with Scientist-Aligned Workflows
Dec 18, 2025Despite advances in scientific AI, a coherent framework for Scientific General Intelligence (SGI)-the ability to autonomously conceive, investigate, and reason across scientific domains-remains lacking. We present an operational SGI definition grounded in the Practical Inquiry Model (PIM: Deliberation, Conception, Action, Perception) and operationalize it via four scientist-aligned tasks: deep research, idea generation, dry/wet experiments, and experimental reasoning. SGI-Bench comprises over 1,000 expert-curated, cross-disciplinary samples inspired by Science's 125 Big Questions, enabling systematic evaluation of state-of-the-art LLMs. Results reveal gaps: low exact match (10--20%) in deep research despite step-level alignment; ideas lacking feasibility and detail; high code executability but low execution result accuracy in dry experiments; low sequence fidelity in wet protocols; and persistent multimodal comparative-reasoning challenges. We further introduce Test-Time Reinforcement Learning (TTRL), which optimizes retrieval-augmented novelty rewards at inference, enhancing hypothesis novelty without reference answer. Together, our PIM-grounded definition, workflow-centric benchmark, and empirical insights establish a foundation for AI systems that genuinely participate in scientific discovery.
ATLAS: A High-Difficulty, Multidisciplinary Benchmark for Frontier Scientific Reasoning
Nov 18, 2025The rapid advancement of Large Language Models (LLMs) has led to performance saturation on many established benchmarks, questioning their ability to distinguish frontier models. Concurrently, existing high-difficulty benchmarks often suffer from narrow disciplinary focus, oversimplified answer formats, and vulnerability to data contamination, creating a fidelity gap with real-world scientific inquiry. To address these challenges, we introduce ATLAS (AGI-Oriented Testbed for Logical Application in Science), a large-scale, high-difficulty, and cross-disciplinary evaluation suite composed of approximately 800 original problems. Developed by domain experts (PhD-level and above), ATLAS spans seven core scientific fields: mathematics, physics, chemistry, biology, computer science, earth science, and materials science. Its key features include: (1) High Originality and Contamination Resistance, with all questions newly created or substantially adapted to prevent test data leakage; (2) Cross-Disciplinary Focus, designed to assess models' ability to integrate knowledge and reason across scientific domains; (3) High-Fidelity Answers, prioritizing complex, open-ended answers involving multi-step reasoning and LaTeX-formatted expressions over simple multiple-choice questions; and (4) Rigorous Quality Control, employing a multi-stage process of expert peer review and adversarial testing to ensure question difficulty, scientific value, and correctness. We also propose a robust evaluation paradigm using a panel of LLM judges for automated, nuanced assessment of complex answers. Preliminary results on leading models demonstrate ATLAS's effectiveness in differentiating their advanced scientific reasoning capabilities. We plan to develop ATLAS into a long-term, open, community-driven platform to provide a reliable "ruler" for progress toward Artificial General Intelligence.
From Supervision to Exploration: What Does Protein Language Model Learn During Reinforcement Learning?
Oct 02, 2025Protein language models (PLMs) have advanced computational protein science through large-scale pretraining and scalable architectures. In parallel, reinforcement learning (RL) has broadened exploration and enabled precise multi-objective optimization in protein design. Yet whether RL can push PLMs beyond their pretraining priors to uncover latent sequence-structure-function rules remains unclear. We address this by pairing RL with PLMs across four domains: antimicrobial peptide design, kinase variant optimization, antibody engineering, and inverse folding. Using diverse RL algorithms and model classes, we ask if RL improves sampling efficiency and, more importantly, if it reveals capabilities not captured by supervised learning. Across benchmarks, RL consistently boosts success rates and sample efficiency. Performance follows a three-factor interaction: task headroom, reward fidelity, and policy capacity jointly determine gains. When rewards are accurate and informative, policies have sufficient capacity, and tasks leave room beyond supervised baselines, improvements scale; when rewards are noisy or capacity is constrained, gains saturate despite exploration. This view yields practical guidance for RL in protein design: prioritize reward modeling and calibration before scaling policy size, match algorithm and regularization strength to task difficulty, and allocate capacity where marginal gains are largest. Implementation is available at https://github.com/chq1155/RL-PLM.
A Survey of Scientific Large Language Models: From Data Foundations to Agent Frontiers
Aug 28, 2025



Scientific Large Language Models (Sci-LLMs) are transforming how knowledge is represented, integrated, and applied in scientific research, yet their progress is shaped by the complex nature of scientific data. This survey presents a comprehensive, data-centric synthesis that reframes the development of Sci-LLMs as a co-evolution between models and their underlying data substrate. We formulate a unified taxonomy of scientific data and a hierarchical model of scientific knowledge, emphasizing the multimodal, cross-scale, and domain-specific challenges that differentiate scientific corpora from general natural language processing datasets. We systematically review recent Sci-LLMs, from general-purpose foundations to specialized models across diverse scientific disciplines, alongside an extensive analysis of over 270 pre-/post-training datasets, showing why Sci-LLMs pose distinct demands -- heterogeneous, multi-scale, uncertainty-laden corpora that require representations preserving domain invariance and enabling cross-modal reasoning. On evaluation, we examine over 190 benchmark datasets and trace a shift from static exams toward process- and discovery-oriented assessments with advanced evaluation protocols. These data-centric analyses highlight persistent issues in scientific data development and discuss emerging solutions involving semi-automated annotation pipelines and expert validation. Finally, we outline a paradigm shift toward closed-loop systems where autonomous agents based on Sci-LLMs actively experiment, validate, and contribute to a living, evolving knowledge base. Collectively, this work provides a roadmap for building trustworthy, continually evolving artificial intelligence (AI) systems that function as a true partner in accelerating scientific discovery.
Position: Intelligent Science Laboratory Requires the Integration of Cognitive and Embodied AI
Jun 24, 2025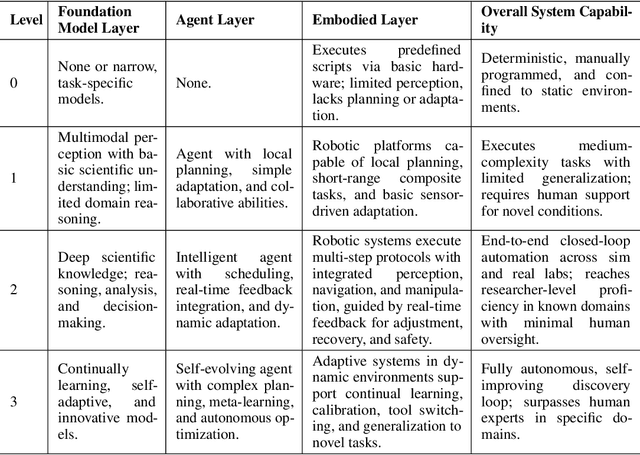
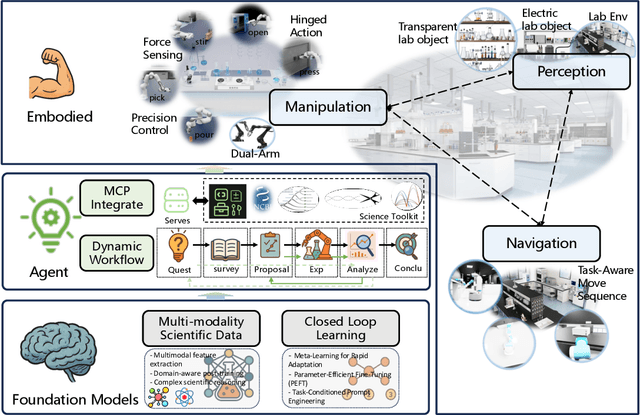
Scientific discovery has long been constrained by human limitations in expertise, physical capability, and sleep cycles. The recent rise of AI scientists and automated laboratories has accelerated both the cognitive and operational aspects of research. However, key limitations persist: AI systems are often confined to virtual environments, while automated laboratories lack the flexibility and autonomy to adaptively test new hypotheses in the physical world. Recent advances in embodied AI, such as generalist robot foundation models, diffusion-based action policies, fine-grained manipulation learning, and sim-to-real transfer, highlight the promise of integrating cognitive and embodied intelligence. This convergence opens the door to closed-loop systems that support iterative, autonomous experimentation and the possibility of serendipitous discovery. In this position paper, we propose the paradigm of Intelligent Science Laboratories (ISLs): a multi-layered, closed-loop framework that deeply integrates cognitive and embodied intelligence. ISLs unify foundation models for scientific reasoning, agent-based workflow orchestration, and embodied agents for robust physical experimentation. We argue that such systems are essential for overcoming the current limitations of scientific discovery and for realizing the full transformative potential of AI-driven science.